
Sodium ferulate prevents amyloid-beta-induced neurotoxicity through suppression of p38 MAPK and upregulation of ERK-1/2 and Akt/protein kinase B in rat hippocampus1
Introduction
Alzheimer disease (AD) is a neurodegenerative disorder characterized by progressive deposition of amyloid-β (Aβ) peptide in the brain to form senile plaques[1]. Neuronal cell loss is one feature of AD. Despite the ambiguity of the effectors of neuron loss in the AD brain, recent reports demonstrate that it is an apoptotic process. Cultured cortical neurons exposed to Aβ1–40 exhibited increased expression of the proapoptotic protein Bax, increased activation of caspase-3 (a marker of apoptotic cell death), and increased terminal deoxynucleodityl transferase-mediated dUTP nick-end labeling reactivity[2]. Neuronal apoptosis is also seen in human AD brain[3]. Activation of the apoptotic cascade induced by Aβ could also explain many of the features of the disease and its progression. Furthermore, the proinflam-matory cytokine, interleukin (IL)-1β, plays a significant role in mediating the effects of Aβ[2]. However, the underlying mechanisms of toxicity and activition of the neuronal cellular signaling cascades induced by Aβ are not fully understood.
It has become increasingly evident that there is a complex balance between survival and apoptotic signaling pathways in neurons that determines whether they will survive or die. For example, the serine/threonine kinase Akt/protein kinase B (PKB) is activated via a phosphoinositide 3-kinase (PI3K)-dependent signaling pathway when cells or tissues are exposed to growth factors, insulin, and certain cytokines[4]. Akt/PKB has received widespread attention as an important anti-apoptotic protein[5]. The extracellular signal-regulated kinase (ERK) pathway plays a major role in regulating cell growth and differentiation[6]. In contrast, the stress-activated protein kinases, including Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), have been proposed to be important signaling components linking extracellular stimuli to cellular responses. JNK and p38 are highly activated in response to a variety of stress signals[7,8]. Their activation is most frequently associated with induction of apoptosis. However, the role of the MAPK and PI3K/Akt pathways in Aβ toxicity is unclear.
Sodium ferulate (SF), extracted from a traditional Chinese herbal medicine, has potent antioxidant[9] and anti-inflammatory activities[10]. It has recently been reported that long-term administration of ferulic acid protects mice against learning and memory deficits induced by centrally administered β-amyloid[11]. The primary site of action of ferulic acid could be microglia[12]. These previous reports prompted us to examine whether SF can suppress the Aβ-induced inflammatory response and neuronal apoptotic death in rat hippocampus. In addition, we considered that increased IL-1β concentration and upregulation of the IL-1β-induced cell signaling cascades might be accompanied by downregulation of survival signals. Therefore, in the present study, we investigated the MAPK and Akt/PKB signaling events in the inflammatory response and apoptosis evoked by preaggregated Aβ25-35, and the protective effect caused by the oral administration of SF in vivo. Aβ25-35, a short synthetic peptide possessing properties similar to Aβ1-40 and Aβ1-42[13,14], is suitable to be used in the study of Aβ toxicity.
Materials and methods
Materials Amyloid-β25-35 (Sigma Chemical Co, St Louis, MO, USA) was resuspended at a concentration of 1 mmol/L. To obtain the aggregated form of Aβ25-35, the peptide solution was placed in an incubator at 37 °C for 48 h. SF, a colorless power with purity >99%, was obtained from Suzhou Changtong Chemical Co (Suzhou, China). Anti-phospho-ERK1/2 (Thr202/Tyr 204), anti-ERK1/2, anti-phospho-p38 MAPK (Thr180/Tyr182), anti-phospho-Akt/PKB (Thr-308), anti-Akt/PKB, and anti-Glial Fibrillary Acidic Protein (GFAP) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-IL-1β, anti-Fas ligand (FasL) and anti-caspase-3 antibodies were purchased from BOSTER Biological Engineering Co (Wuhan, China). Goat anti-rabbit and goat anti-mouse secondary antibodies were obtained from Santa Cruz Biotechnology. RNA polymerase chain reaction (PCR) kit (AMV) was purchased from TaKaRa Biotechnology (Dalian, China). O-Dianisidine tetrazotited and β-naphthyl acid phosphate were purchased from Sigma Chemical Co. The caspase-3 colorimetric assay kit was obtained from BD Bioscience Clontech (1020 East Meadow Circle Palo Alto, CA).
Animals and drug treatment Groups of Sprague Dawley rats (Grade II, certificate N
Immunohistochemical staining for glial fibrillary acidic protein Seven days after injection of Aβ, the rats were perfused transcardially with 4% paraformaldehyde in phosphate-buffered saline (PBS). The brains were removed and post-fixed for 24 h and were embedded in paraffin wax. Serial coronal sections (5 µm thickness) were cut from various sections of the brain. After the coronal sections were rinsed in PBS 3 times, endogenous peroxidase activity was blocked by incubation with 3% H2O2 for 10 min. The sections were incubated with 10% normal goat serum. After the blocking serum was removed, sections were incubated with polyclonal antibody against GFAP (1:100 in Tris-buffered saline (TBS) containing 2.5% normal goat serum) overnight at 4 °C, then with biotinylated secondary antibody at 37 °C for 20 min. The GFAP-positive cells were detected using strept-avidin-biotin complex (SABC) and DAB kits.
Western blot analysis Western blotting was carried out for the analysis of IL-1β, p38 MAPK, ERK1/2, Akt/PKB, FasL and caspase-3. Hippocampal CA1 areas were stereotaxically dissected free. The tissue was homogenized in RIPA buffer [1% Triton, 0.1% sodium dodecylsulfate (SDS), 0.5% deoxycholate, ethylenediaminetetracetic acid 1 mmol/L, Tris 20 mmol/L (pH 7.4), NaCl 150 mmol/L, NaF 10 mmol/L], and insoluble material was removed by centrifugation at 12 000×g for 20 min at 4 °C. Protein concentrations were quantified by the method of Lowry [15]. Tissue samples were equalized for protein concentration. Proteins were resolved by 9%–15% SDS-polyacrylamide gel electrophoresis (PAGE), and transferred to nitrocellulose membranes. The membranes were blocked with 5% milk powder in TBS (pH 7.6) for 1 h and incubated overnight at 4 °C with suitably diluted primary antibodies. After extensive washing with TBS, the membranes were incubated with anti-IgG-alkaline phosphatase conjugate. Finally, the blots were developed with the alkaline phosphatase substrate O-dianisidine tetrazotited along with β-naphthyl acid phosphate. Quantification of protein bands was achieved by densitometric analysis using Chem Image 5500 software (UVP, USA).
Analysis of caspase-3 activity The activity of caspase-3 proteases was measured using a caspase-3 colorimetric activity assay kit. Briefly, hippocampal CA1 areas were washed, homogenized in lysis buffer, incubated on ice for 20 min, analyzed for protein concentration, and diluted to equalize for protein concentration. All samples (95 µL) were added to 5 µL of 1 mmol/L caspase-3 substrate (Ac-DEVD-pNA, 50 µmol/L final concentration) and incubated for 1 h at 37 °C. Cleavage of Ac-DEVD-pNA by active caspase-3 resulted in the liberation of p-nitroanilide (pNA) into solution. The release of pNA was quantitated spectrophotometrically by measuring absorbance at 405 nm (Biocell 2010, Anthos Labtec Instruments, Austria), and enzyme activity was calculated with reference to a standard curve of pNA concentration versus absorbance. The data were represented as the nmol pNA·min-1·mg of protein-1.
Polymerase chain reaction analysis for interleukin-1β gene expression A semiquantitative reverse transcription-PCR (RT-PCR) assay was used to determine the mRNA levels of IL-1β in relation to β-actin message. Total RNA was extracted from hippocampal CA1 areas using TRIzol reagent (Invitrogen Life Technologies, Paisley, PA4 9RF, UK). cDNA synthesis was carried out on 1 mg total RNA using random 9mers by RT with 2.5 IU of Avian Myeloblastosis Virus (AMV) reverse transcriptase in RT buffer in the presence of 1 mmol/L each of dNTP and 10 IU of RNase inhibitor. The thermal cycler (Biometra, Germany) was programmed for 10 min at 30 °C, 30 min at 42 °C, 5 min at 99 °C, and 5 min at 5 °C. Equal amounts of cDNA were used for PCR amplification for 35 cycles, using a 3-step program (30 s at 94 °C, 30 s at 56 °C, and 1 min at 72 °C). After amplification, the products were separated by 7.5% SDS-PAGE (cast in the presence of ethidium bromide) and visualized under UV light. The following sequences of the primers were used: rat IL-1β (sense), 5'-TGA CTC GTG GGA TGA TGA CG-3'; rat IL-1β (antisense), 5'-CTG GAG ACT GCC CAT TCT CG-3'; β-actin (sense), 5'-GTG GGC CGC TCA AGG CAC CAA-3'; β-actin (antisense), 5'-CTT TAG CAC GCA CTG TAG TTT CTC-3'.
Statistical analysis All data were presented as mean±SD. Statistical analysis was carried out with one-way ANOVA, followed by LSD’s post hoc test, which was provided by SPSS 11.5 statistical software. The level of significance was accepted as P<0.05.
Results
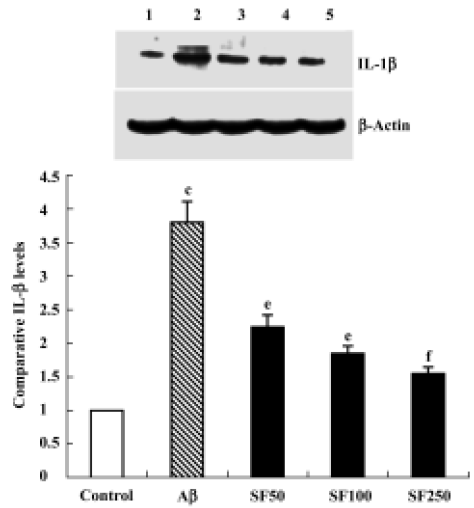
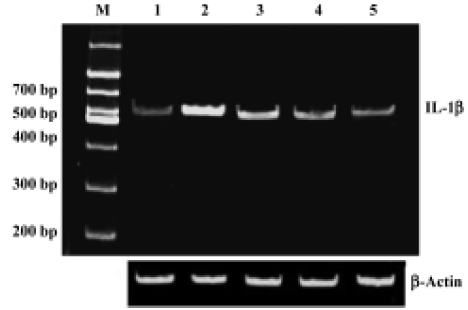
Sodium ferulate inhibited the amyloid-β25-35-induced increase in interleukin-1β protein synthesis and mRNA expression Intracerebroventricular injection of preaggregated Aβ25-35 increased protein expression of IL-1β, and densitometric analysis revealed that the mean value in samples prepared from Aβ-treated rats was significantly higher than that of control rats. SF (50, 100, and 250 mg/kg) significantly inhibited a Aβ25-35-induced increase in protein expression of IL-1β in a dose-dependent manner (Figure 1). An Aβ-associated increase in IL-1β protein was mirrored by an Aβ-induced increase in IL-1β mRNA levels. SF induced a similar inhibition in IL-1β mRNA (Figure 2). On the other hand, the control results showed that only SF (100 mg/kg, daily for 4 weeks) had not effect on basal levels of IL-1β protein and mRNA expression in hippocampal CA1 areas (data not shown).
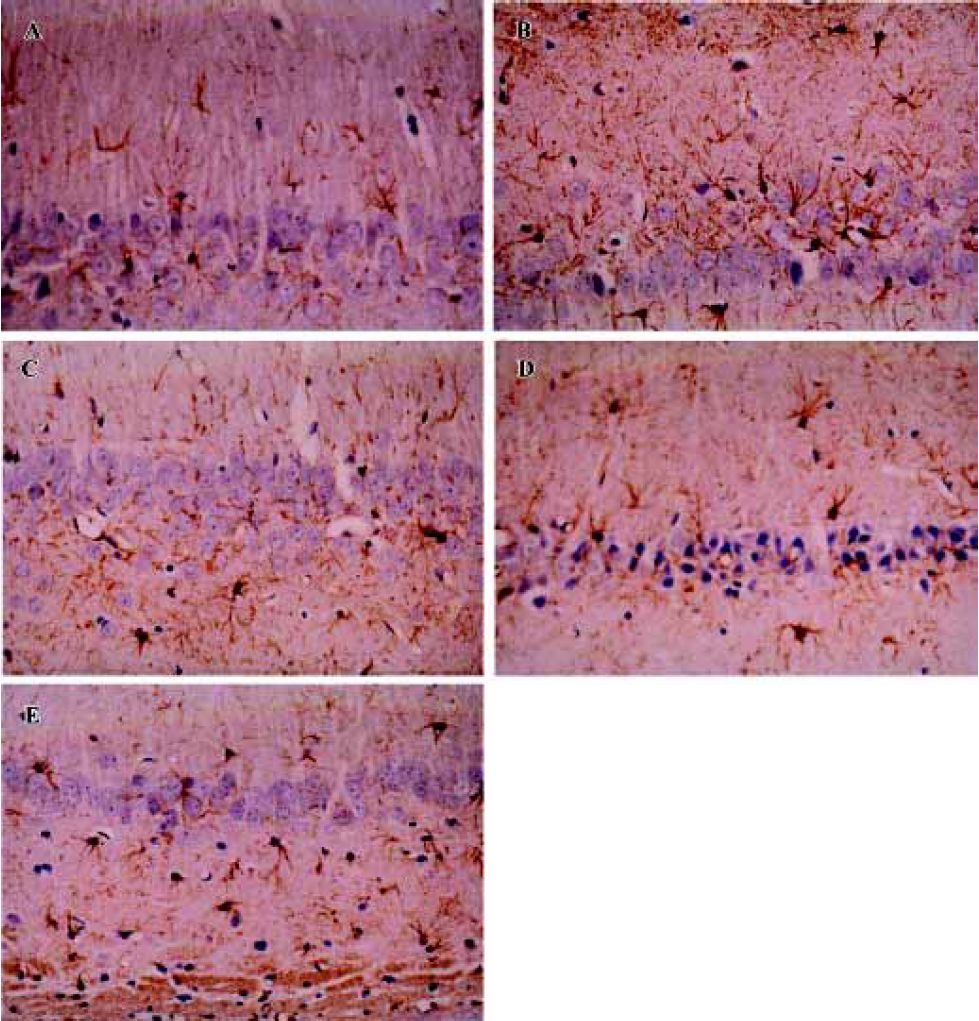
Sodium ferulate inhibited the amyloid-β25-35-induced astrocyte activation The astrocyte reaction was visualized by means of the immunoreactivity for glial fibrillar acidic protein, a specific marker of astrocytes. Aβ25-35 resulted in infiltration of astrocytes in hippocampal CA1, as well as transformation of astrocytes from a resting to an activated state, highlighted by phenotypic changes characterized by long, thick branching. SF at 50 mg/kg, 100 mg/kg, and 250 mg/kg significantly inhibited the Aβ25-35-induced astrocytic reaction in hippocampal CA1 (Figure 3).
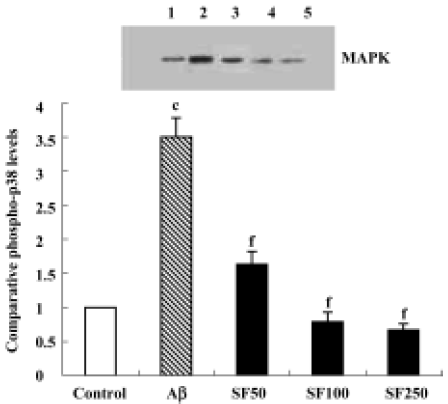
Sodium ferulate inhibited the amyloid-β25-35-induced increase in phospho-p38 MAPK expression The p38 MAPK pathway, a major proinflammatory signal transduction pathway, is hyperactivated in the AD brain[16]. Therefore, p38 MAPK activation was examined by immunoblotting in rat hippocampal CA1 using anti-phospho-p38 MAPK, an antibody that specifically recognizes the activated, tyrosine-phosphorylated form of p38 MAPK. The results showed that Aβ25-35 led to a significant increase in phospho-p38 MAPK protein expression. The Aβ-induced increase in activation of p38 MAPK was paralleled by an Aβ-induced increase in IL-1β protein expression. The Aβ-induced increase in activation of p38 MAPK was prevented by SF (50, 100, and 250 mg/kg, daily for 4 weeks) in a dose-dependent manner (Figure 4). In addition, SF (100 mg/kg, daily) treatment alone for 4 weeks did not result in a significant reduction in the basal expression of phospho-p38 MAPK in rat hippocampus (data not shown).
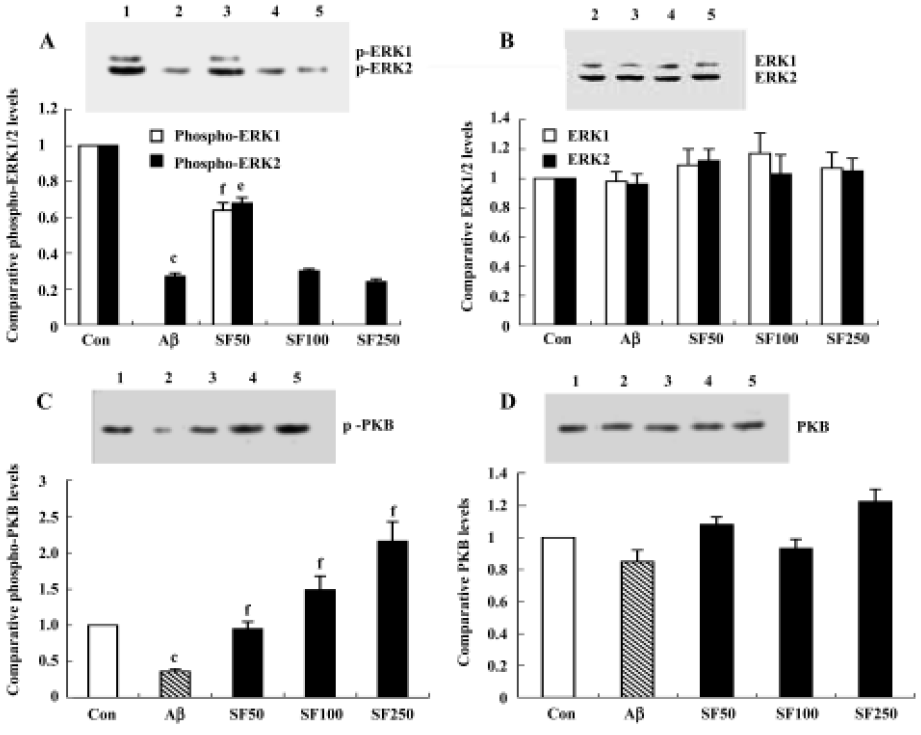
Intracerebroventricular injection of amyloid-β25-35 downregulated ERK-1/2, Akt/protein kinase B and the effects of sodium ferulate Although IL-1β-induced enhancement of p38 MAPK activation may lead to deterioration in cell function and even cell death, we considered that downregulation of cell survival signals may also contribute to Aβ-induced damages and, therefore, we analyzed the activity of ERK1/2 and Akt/PKB in the hippocampal CA1 region. The results showed that 7 d after injection, Aβ25-35 elicited a significant decrease in the activated, tyrosine-phosphorylated form of ERK1 and ERK2, especially phospho-ERK1, compared to control rats. Aβ completely inhibited the expression of phospho-ERK1. SF (50 mg/kg, daily for 4 weeks) partly abolished the Aβ25-35-induced decrease in ERK1 and ERK2 activation, but SF (100 mg/kg and 250 mg/kg, daily for 4 weeks) did not prevent the decrease in phosphorylated ERK1 and ERK2 induced by Aβ25-35 (Figure 5A). SF treatment alone (100 mg/kg, daily for 4 weeks) exerted a decrease in phosphorylated ERK1 and ERK2 that did not reach statistical significance compared with control rats (data not shown). However, the underlying cause of this effect of SF (100 mg/kg and 250 mg/kg) on phosphorylated ERK1 and ERK2 remains to be identified. No significant difference in total ERK1/2 was apparent, as shown in the sample immunoblot (Figure 5B). Treatment with Aβ25-35 also decreased the expression of phosphorylated Akt/PKB compared with control rats. SF (50 mg/kg, 100 mg/kg and 250 mg/kg, daily for 4 weeks) completely reversed the effect of Aβ25-35 on phosphorylated Akt/PKB compared with control rats in a dose-dependent manner. The level of phospho-Akt/PKB expression in Aβ25-35+ SF (100 mg/kg and 250 mg/kg) was significantly greater than in control rats (Figure 5C). However, no significant difference in the expression of total Akt/PKB was observed among treatment groups (Figure 5D). In addition, SF alone (100 mg/kg, daily for 4 weeks) had no obvious effect on the basal phosphorylation of Akt/PKB in the hippocampus (data not shown).
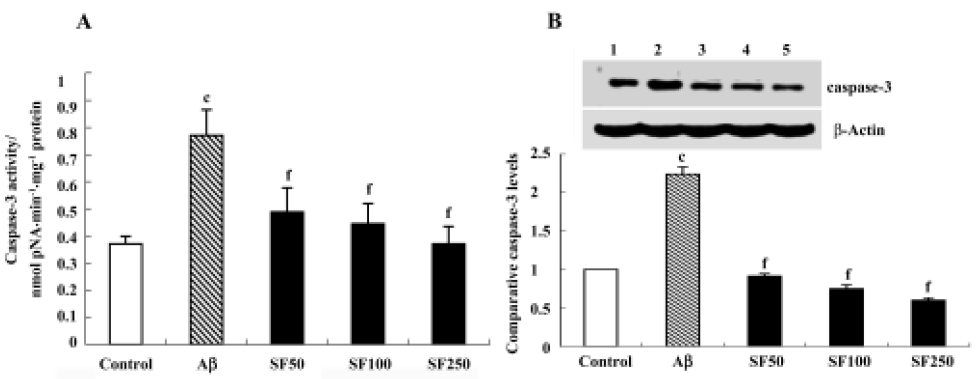
Sodium ferulate prevented the amyloid-β25-35-induced increase in caspase-3 activity and caspase-3 protein expression in rat hippocampus Caspase-3 activity was measured by cleavage of the caspase-3 substrate (Ac-DEVD-pNA). As shown in Figure 6A, caspase-3 activity was significantly enhanced in hippocampal CA1 prepared from Aβ-treated rats compared with control rats. This Aβ-induced increase in caspase-3 activity was inhibited by SF (50 mg/kg, 100 mg/kg and 250 mg/kg). Western blot analysis showed that Aβ25-35 evoked a significant increase in caspase-3 protein expression. SF (50 mg/kg, 100 mg/kg and 250 mg/kg) prevented the increase in Aβ-induced caspase-3 protein expression (Figure 6B).
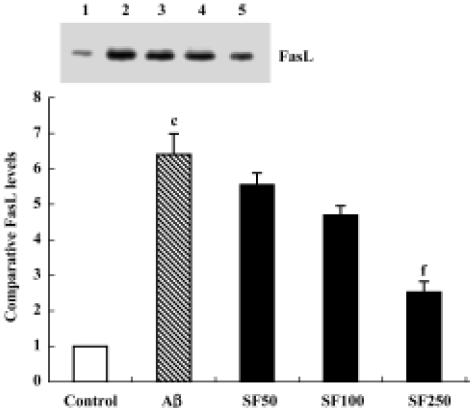
Sodium ferulate decreased the amyloid-β25-35-induced expression level of Fas ligand protein Previous studies have shown that survival factor withdrawal leads to the induction of FasL protein and mRNA in cerebellar guanule neurons and PC 12 cells[17]. The binding of FasL to the Fas receptor is a prototypic signal for apoptosis, and therefore it was of interest to determine whether SF inhibits the Aβ-induced increase in FasL protein expression. As shown in Figure 7, Aβ25-35 significantly enhanced the expression level of FasL protein in hippocampal CA1 regions. SF (50 mg/kg, 100 mg/kg and 250 mg/kg) demonstrated inhibition of the Aβ-induced increase in FasL protein expression level in a dose-dependent manner (Figure 7).
Discussion
Injection of aggregated Aβ into the brain of experimental animals may represent a valuable tool for studying the neurotoxic effect of this peptide. We found that intracerebro-ventricular injection of Aβ25-35 induced an increase in IL-1β protein and mRNA expression in hippocampal tissue, and this increase, in combination with enhanced activation of p38 MAPK and reduced activation of ERK1/2 and Akt/PKB, mediates the Aβ-induced activation of cell death events as assessed by activation of caspase-3. However, several of these changes, including Aβ-induced increase in caspase-3 activation, were prevented by treatment with SF. Indeed in AD brain, the increased levels of phosphorylated (active) p38 MAPK and diminished expression of Akt/PKB were detected[16,18]. Maher et al also found that increased IL-1β in the cortex of aged rats was accompanied by downregulation of ERK and PI3 kinase (an upstream kinase of Akt/PKB)[19]. These observations bear a marked similarity to our results. The Aβ-induced diminished expression of phospho-ERK1/2 and phospho-Akt/PKB in hippocampal tissue indicates that the neuron survival signal transduction pathway is impaired. The Aβ-induced increase in p38 MAPK phosphorylation and decrease in phosphorylated ERK1/2 and Akt/PKB parallel some changes that are hallmarks of cell death. For example, an increase in caspase-3 activity and FasL protein expression were observed in hippocampal tissue treated with Aβ, suggesting that sequential activation triggers apoptotic changes in the hippocampus.
Increased activation of p38 MAPK accompanied the Aβ-induced increase in IL-1β concentration, consistent with previous observations in hippocampal cells[20]. The evidence presented here pinpoints IL-1β-induced increased activation of p38 MAPK as a pivotal event in triggering changes that are characteristic of apoptotic cell death, for example, caspase-3 activation. Thus, inhibition of p38 MAPK by SB203580 has been shown to prevent the increase in IL-1β-induced caspase-3 activity[21]. A significant finding of this study is that SF treatment prevents the Aβ-induced increase in activation of p38 MAPK, caspase-3 and FasL expression, indicating a potential neuroprotective effect of SF. These findings suggest that the effects of SF on the activity of p38 MAPK and caspase-3 may be secondary to its ability to suppress the Aβ-induced increase in IL-1β synthesis.
It has also been shown that cell death is accompanied by a decrease in survival signals[22]. PI3K/Akt and ERK1/2 have been shown to be important for neuronal survival. Over-expressing active Akt/PKB rescues cells from apoptosis[23]. We report in this study that, in addition to increased p38 MAPK activation, the increase in casepase-3 activation may be associated with attenuated activity of ERK and Akt/PKB. SF treatment significantly prevented these Aβ-induced changes and, therefore, in Aβ-treated rats that were treated with SF, we found that caspase-3 activity was significantly decreased and paralleled the changes in activities of both ERK and Akt/PKB. These results are consistent with those described by Daniels et al[24] and Meng et al[18]. Therefore, downregulation of Akt/PKB may be another mechanism by which Aβ induces apoptosis. To our knowledge, this is the first indication that Akt/PKB activation in the hippocampus is decreased by Aβ in vivo. Thus, Aβ-induced increase in caspase-3 activity, which is considered to be a reliable indicator of apoptotic cell death, may arise from the coupled increase in p38 MAPK activation and decrease in ERK and Akt/PKB activation.
Several observations have contributed to the development of the idea that FasL expression and, consequently, Fas activation play a role in neurodegeneration[2,25], and we report that increased hippocampal expression of FasL accompanied the Aβ-induced increase in p38 MAPK phosphorylation and caspas-3 activation. The binding of Fas to FasL triggers activation of caspase-8 and, in turn, caspase-8 activates caspase-3[26]. SF markedly prevented the Aβ-induced increase in FasL protein expression.
In conclusion, our data suggest that Aβ-induced increased IL-1β, coupled with increased p38 MAPK activation, leads to cell death as assessed by activation of caspase-3. In addition, the data also show that downregulation of the survival signals ERK and Akt/PKB may contribute to the demise of the cells. These are significantly abrogated by SF treatment, which also attenuates Aβ-induced increase in caspase-3 activity and FasL expression.
References
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18:351-7.
- Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, et al. Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-β on long term potentiation and cell death in hippocampus. A role for interleukin-1β? J Biol Chem 2003;278:27971-80.
- Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellnger K, et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am J Pathol 1999;155:1459-66.
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science 1997;275:628-30.
- Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol 2000;151:483-94.
- Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol 1997;9:180-6.
- Wang X, Martindale JL, Liu Y, Holbrook NJ. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem J 1998;333:291-300.
- Namgung U, Xia Z. Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N-terminal protein kinase 3 and p38 mitogen-activated protein kinase. J Neurosci 2000;20:6442-51.
- Scott BC, Butler J, Halliwell B, Aruoma OI. Evaluation of the antioxidant action of ferulic acid and catechins. Free Radic Res Commun 1993;19:241-53.
- Fernandez MA, Saenz MT, Garcia MD. Anti-inflammatory activity in rats and mice of phenolic acids isolated from Scrophularia frutescens. J Pharm Pharmacol 1998;50:1183-6.
- Yan JJ, Cho JY, Kim HS, Kim KL, Jung JS, Huh SO, et al. Protection against beta-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br J Pharmacol 2001;133:89-96.
- Kim HS, Cho JY, Kim DH, Yan JJ, Lee HK, Suh HW, et al. Inhibitory effects of long-term administration of ferulic acid on microglial activation induced by intracerebroventricular injection of β-amyloid peptide (1–42) in mice. Biol Pharm Bull 2004;27:120-1.
- Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem 1995;64:253-65.
- Saito T, Kijima H, Kiuchi Y, Isobe Y, Fukushima K. β-Amyloid induces caspase-dependent early neurotoxic change in PC12 cells: correlation with H2O2 neurotoxicity. Neurosci Lett 2001;305:61-4.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951;193:265-75.
- Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, et al. p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem 1999;72:2053-8.
- Le-Niculescu H, Bonfoco E, Easuya Y, Claret F-X, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol 1999;19:751-63.
- Meng Y, Xu H, Wang R, Ji Z, Yu S, Zhou J, et al. Impairment of signal transduction pathway on neuronal survival in brain of Alzheimer’s disease. Chin J Pathol 2002;31:502-5.
- Maher FO, Martin DS, Lynch MA. Increased IL-1β in cortex of aged rats is accompanied by downregulation of ERK and PI-3 kinase. Neurobiol Aging 2004;25:795-806.
- O’Donnell E, Vereker E, Lynch MA. Age-related impairment in LTP is accompanied by enhanced activity of stress-activated protein kinases: analysis of underlying mechanisms. Eur J Neurosci 2000;12:345-52.
- Martin DS, Lonergan PE, Boland B, Fogarty MP, Brady M, Horrobin DF, . Apoptotic changes in the aged brain are triggered by interleukin-1β-induced activation of p38 and reversed by treatment with eicosapentaenoic acid. J Biol Chem 2002; 277: 34 239–46.
- Macdonald NJ, Decorti F, Pappas TC, Taglialatela G. Cytokine/neurotrophin interaction in the aged nervous system. J Anat 2000;197:543-51.
- Liot G, Gabriel C, Cacquevel M, Ali C, MacKenzie ET, Buisson A, et al. Neurotrophin-3-induced PI-3 kinase/Akt signaling rescues cortical neurons from apoptosis. Exp Neurol 2004;187:38-46.
- Daniels WM, Hendricks J, Salie R, Taljaard JJ. The role of the MAP-kinase superfamily in beta-amyloid toxicity. Metab Brain Dis 2001;16:175-85.
- Su JH, Anderson AJ, Cribbs DH, Tu C, Tong L, Kesslack P, et al. Fas and Fas ligand are associated with neuritic degeneration in the AD brain and participate in beta-amyloid-induced neuronal death. Neurobiol Dis 2003;12:182-93.
- Suda T, Nagata S. Purification and characterization of the Fas-ligand that induces apoptosis. J Exp Med 1994;179:873-9.