
Hypoxia reoxygenation induces premature senescence in neonatal SD rat cardiomyocytes1
Introduction
Cellular senescence has been defined by Hayflick and Moorehead as the ultimate and irreversible loss of replicative capacity occurring in primary somatic cell culture[1]. Senescence was generally studied in culture, different markers of the senescent phenotype, as mitochondrial dehydration[2], senescence-associated β-gal activity[3], and senescence-associated gene expression[4,5]. Senescence has been identified in human fibroblasts[6], keratinocytes[7], endothelial cells[8], smooth muscle cells[9], tumor cells[10], hematopoietic cells[11], and cardiomyocytes[12,13].
The mammalian heart is an obligate aerobic organ. Oxygen, beyond its indispensable role in cardiac energy metabolism, plays a central role in other biological processes that can be determinants of cardiac function, including the determination of cardiac gene expression patterns[14]. The possibility that cardiac aging is an independent determinant of morbidity and mortality has faced opposition, and emphasis has been placed on age-associated changes, which increase the chances of cardiovascular events in the elderly. Treatment of cardiac diseases in elderly patients has resulted in a prolongation of the average lifespan. However, the maximum lifespan has not increased in the last 70 years[15], suggesting that cellular aging may play a more important role than generally expected.
Accumulating evidence suggests that telomerase plays a role in cellular senescence in vitro and in vivo[16,17]. The enzyme telomerase, a ribonucleoprotein, is crucial for maintaining telomere length. Telomerase reverse transcriptase (TERT) is an important component of telomerase. It can counteract telomere shortening and prevent senescence[18]. Senescence in human fibroblasts can be overcome by over-expression of TERT[19]. Telomerase elongates short telomere, and the cells become immortalized.
Oxidative stress has been implicated in aging and numerous diseases[20,21]. Nevertheless, whether premature senescence could be induced and blocked by TERT in neonatal Sprague-Dawley (SD) rat cardiomyocytes exposed to hypoxia reoxygenation remains unknown. In this study, we investigated the effect of hypoxia reoxygenation on neonatal SD rat cardiomyocytes.
Materials and methods
Experimental animals All experiments were performed in accordance with the protocols approved by Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals of Nanjing Medical University, China.
Cardiomyocytes culture Neonatal SD rat hearts (1-2 d old) were removed under sterile conditions and washed 3 times in phosphate buffered saline (PBS) to remove excess blood cells. The ventricles were minced to small fragments and then agitated gently in a solution of 0.1% trypsin. The mixture was centrifuged at 1000×g for 15 min. The supernatant phase was discarded, and the cells were resuspended in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 100 U/mL of penicillin and 100 µg/mL of streptomycin. Isolated cells were cultured in a flask for 1 h at 37 °C in order to remove fibroblasts. The cultures were incubated in a humidified atmosphere of 5% CO2 and 95% air at 37 °C.
Hypoxia reoxygenation The suspension of cells was diluted to 1.0?106 cells/mL, and 1.5 mL was seeded in 6-well plastic plate and cultured for 72 h. Then, the control cultures were continuously incubated in 5% CO2 and 95% air at 37 °C. The hypoxic cultures were placed within a modular incubator chamber (BioSpherix, Redfield, NY, USA), filled with 1% O2, 5% CO2, and balance N2 for 6 h. The reoxygenated cultures were subjected to 1% O2 and 5% CO2 for 6 h, then 21% oxygen for 4, 8, 12, 24, and 48 h, respectively.
Electron microscopy For electron microscopy, cardio-myocytes were digested with 0.1% trypsin, centrifuged at 1000×g for 10 min and fixed in 2.5% glutaraldehyde; they were postfixed in 1% osmium tetroxide in 0.1 mol/L phosphate buffer, dehydrated through a graded series of ethanol and propylene oxide, and then embedded in epoxy resin. Ultrathin sections of cardiomyocytes were stained with uranyl acetate and observed using an electron microscopy (JEM-1 010, JEOL, Tokyo, Japan).
Cell proliferation assay Cell proliferation was determined using bromodeoxyuridine (BrdU) (Sigma, St Louis, MO, USA) labeling. Experimental cells were pretreated with 10 祄ol/L BrdU for 30 min before hypoxia treatment and continued until time of hypoxia; normal cells were incubated with BrdU with an equivalent dose and time. The labeled cells were observed by immunocytochemistry using a mouse monoclonal antibody directed against BrdU 1:200 (Sigma, USA) and rabbit anti-mouse IgG as the secondary reagent. The labeled cells were viewed with the high-resolution Pathological Image Analysis System 1000 (High Resolution Pathological Image and Word Analysis System, Beijing, China). Stained BrdU and non-stained cells were counted in randomly selected areas on the sections to a total number of 500 cells per fish.
RNA isolation and real-time quantitative PCR Total RNA was isolated using the Trizol (Invitrogen, Life Technologies, Carlsbad, California, USA) method from the cells. RNA was treated with DNase to prevent contamination of genomic DNA. Total RNA (2 µg) was subjected to reverse transcription using reverse transcriptase (TaKaRa Biotechnology Co Ltd, Dalian, China) as described previously[22]. Expression levels of p16INK4a and TERT mRNA were quantified by real-time quantitative PCR, using the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster city, CA, USA). With the use of standard curves, the amount of copy numbers of target genes in each sample was calculated and expressed as a ratio to the β-actin. Primers and probes of target genes were designed using Primer Express software (Applied Biosystems, USA) and they are listed in Table 1.
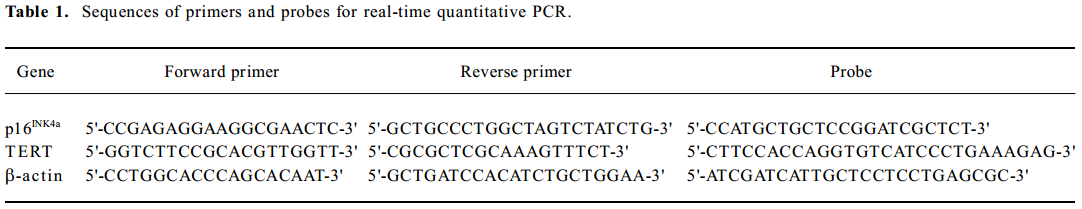
Full table
β-galactosidase staining Cell cultures were washed twice in PBS (pH 7.4) and fixed for 10 min at room temperature with 20% formaldehyde and 2% glutaraldehyde in PBS. After 3 washes in PBS, the cultures were incubated for 24 h at 37 °C in freshly prepared SA-β-galactosidase staining solution containing 1 mg/mL 5-bromo-4-chloro-3-indolyl-β-D-galacto-pyranoside [X-gal; Sigma, USA (pH 6.0)] 400 mmol/L potassium ferrocyanide, 400 mmol/L potassium ferricyanide, and 200 mmol/L MgCl2. The percent of SA-β-galactosidase positive cells was determined by counting the number of blue cells under bright field illumination, and the total number of cells in the same field under phase as a contrast.
Immunohistochemical procedures Slide chambers with cardiomyocytes were treated in 0.5% H2O2 for 5 min and washed in PBS. Non-specific binding was blocked with normal goat serum. Free-floating sections were incubated for 2 h at 37 °C in monoclonal antibodies myosin light chain antibody (Sigma, USA) and TERT (Acris, Hiddenhausen, Germany), all diluted at 1:100 in PBS. The sections were then washed in PBS and incubated for 30 min at 37 °C with the second antibody goat anti-mouse IgG. The protein contents were analyzed using the high-resolution Pathological Image Analysis System 1000.
Telomerase activities assay Cardiomyocytes were washed in PBS, and the pellet was lysed with 200 µL lysis buffer for 30 min at 4 °C as previously described[23]. The proteins were centrifuged for 20 min at 16 000×g, and protein concentrations were determined in the supernatant using the Bradford assay. Telomerase activities were measured with 2 µg protein by the Telo TAGGG Telomerase PCR ELISAplus kit according to the manufacturer's instructions (Roche Molecular Biochemicals, Mannheim, Germany).
Statistical analysis Experiments were performed at least 3 times. Data were expressed as mean±SD. The t test and χ2 were used to determine significant differences between 2 groups. P values less than 0.05 were considered to be significant.
Results
Identification of isolated cells as cardiomyocytes No spindle-shaped fibroblast could be seen. Immunocytochemical analysis revealed that most (>95%) cells in the isolated cultures were positive for sarcomeric myosin light chain (Figure 1).
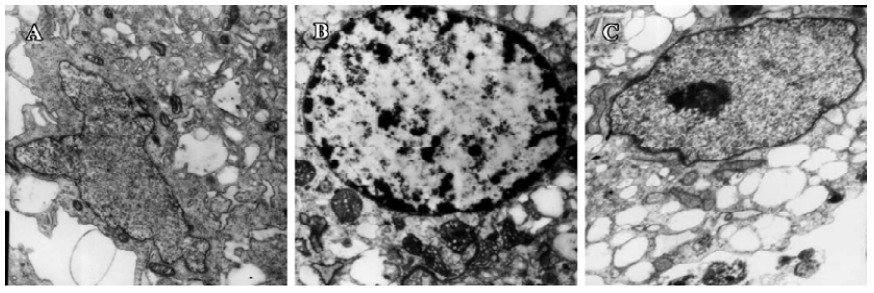
Electron morphology of cardiomyocytes Cardiomyocytes showed normal morphology and equal nuclear chromatin distribution in the control group (Figure 2A). Most cells appeared to have cytoplasmic concentration, nuclear chromatin condensation and margination in the hypoxia group (Figure 2B). The cells approached mitochondrial dehydration with enlarged, flattened nuclear morphology in the hypoxia reoxygenation group (Figure 2C).
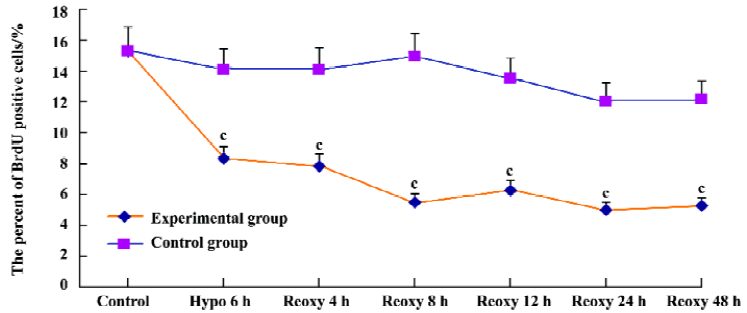
Cell proliferation assay We examined cardiomyocyte proliferation in the hypoxia reoxygenation-treated group by measuring BrdU incorporation. The proportion of BrdU positive cells was 15.32% in the control group. It was reduced significantly in the hypoxia reoxygenation-treated group (P<0.01; Figure 3). These results indicated that cardiomyo-cyte proliferation could be blocked by hypoxia reoxygenation.
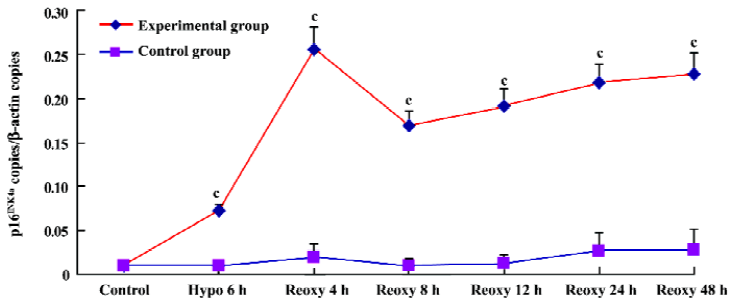
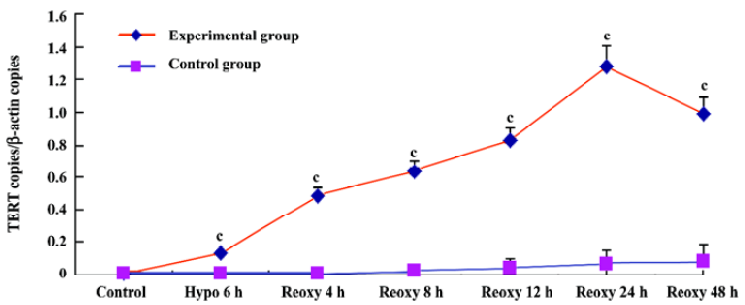
p16INK4a and TERT mRNA expression We investigated whether hypoxia and reoxygenation treatment altered the expression of the p16INK4a and TERT. With the use of real-time quantitative PCR, significant increases in p16INK4a and TERT mRNA levels were observed in the hypoxia reoxygenation-treated group compared with the untreated group (P<0.01), respectively (Figures 4, 5).
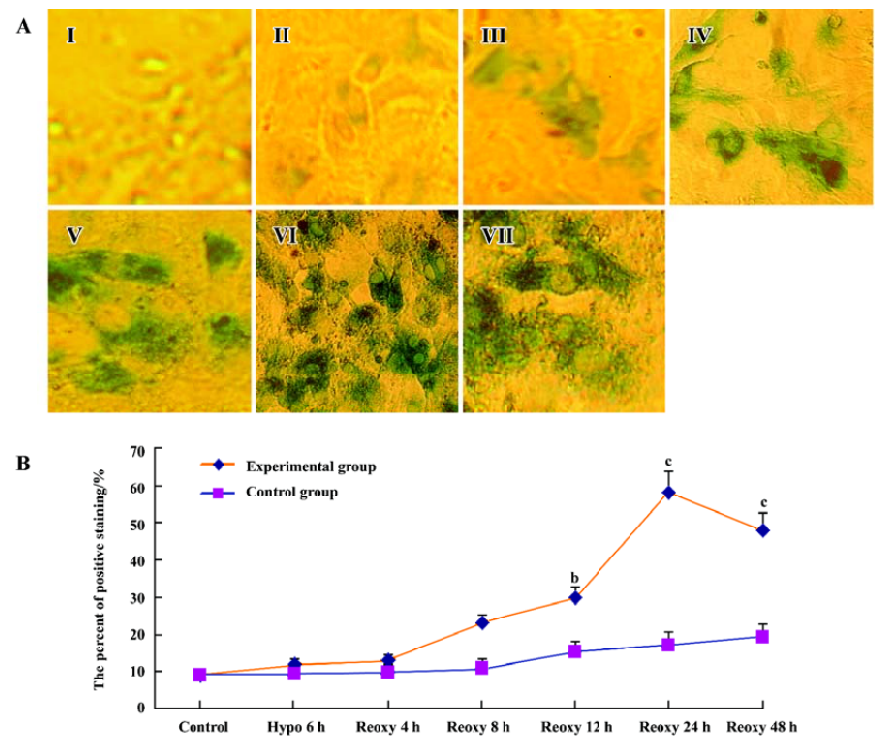
β-galactosidase staining We investigated whether the hypoxia reoxygenation-treated cells were β-galactosidase-positive. With the use of a senescence β-galactosidase staining kit, a small part of the cells were positive for β-galactosidase in the control group, the hypoxia 6 h group, and the reoxygenation 4 and 8 h group, but β-galactosidase-positive cells significantly increased in the reoxygenation 12, 24, and 48 h group (P<0.05 and P<0.01; Figure 6A, 6B).
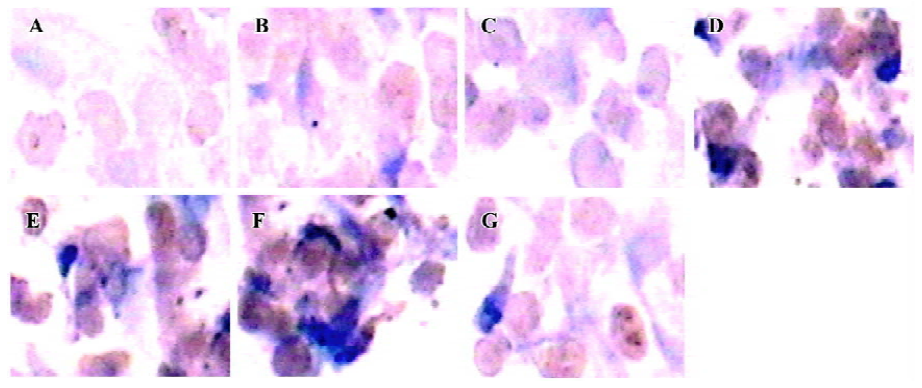
TERT protein In our study, TERT protein expression was determined by immunohistochemistry. The immunohistochemical results showed that the positive cardiomyocytes were stained brown in the intranucleus and cytoplasm; the degree of color in each group was different. TERT protein expression in the group of hypoxia 6 h, reoxygenation 4, 8, 12, 24, and 48 h was significantly increased compared with control group (P<0.01; Figure 7).
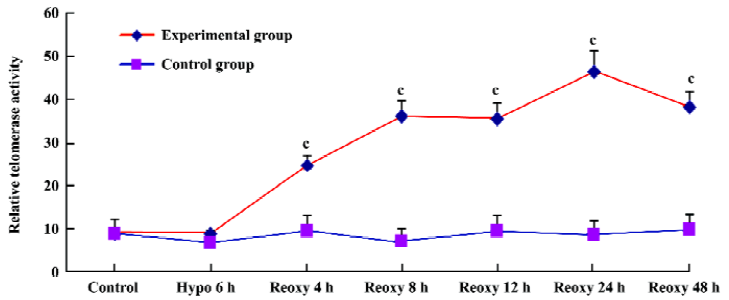
Telomerase activity assay With use of the Telo TAGGG Telomerase PCR ELISAplus kit, relative telomerase activity increased significantly in the hypoxia reoxygenation-treated group compared with the control group (P<0.01; Figure 8).
Discussion
In our study, most cardiomyocytes demonstrated cytoplasmic concentration, nuclear chromatin condensation and margination (Figure 1B), then chromatin gathered at the center of cell nucleus in the hypoxia reoxygenation-treated group (Figure 1C). Mitochondrial dehydration, enlarged, flattened nuclear morphology could be observed in the reoxygenation-treated group (Figure 1C). The results were in accordance with a previously published study[24].
Unlike other cells, the proliferation of cardiomyocytes is so limited after birth; the rate was only 15.32% in the control group. It was reduced significantly with hypoxia reoxygena-tion treatment (Figure 3). At or around birth, cardiomyocytes nearly lose their ability to divide. Cardiomyocyte DNA synthesis is associated with cell proliferation during fetal life, and a second DNA synthesis phase occurring after birth (up to approximately neonatal d 3) is associated only with binucleation[25]. After birth, cardiac growth involves increasing the size of the myocytes without substantial increases in cell number.
p16INK4a mRNA copies in the hypoxia reoxygenation-treated cells were significantly increased compared with the untreated cells (Figure 4). p16INK4a, an inhibitor of the cell cycle[26], encoded potent tumor suppressor proteins[27], and was recognized as the key regulator of premature senescence. Its expression levels were found to increase dramatically in premature senescent cells[28] whose overexpression in mammalian cells could induce G1 phase arrest[29].
β-galactosidase activity has recently been used as a histochemical marker of premature senescence in human fibroblasts, keratinocytes, endothelial and smooth muscle cells[30-32]. In our study, few cardiomyocytes were stained for β-galactosidase in the control group, the hypoxia 6 h group, and the reoxygenation 4 and 8 h group, but the staining intensity increased significantly in the reoxygenation 12, 24, and 48 h group (Figure 6A, 6B). Our findings are similar with a previous study on bone marrow cells[21]. β-Galactosidase activity also correlated with replicative capacity in human mammary epithelial cells[32]. It provided a simple assay for chromosomes that induced senescence[33]. β-Galactosidase activity increased in combination with the expression of senescence-associated proteins p53 and p21WAF1 as well[34].
TERT mRNA and protein expression, relative telomerase activities, were all significantly increased in the hypoxia reoxygenation-treated cells compared with the untreated cells (Figures 5, 7, 8), but they could not inhibit senescence. It has been previously reported that the exposure of human cells to hypoxic conditions results in the induction of telomerase activity through human telomerase reverse transcriptase (hTERT) expression[35,36]. TERT mRNA levels were reported to correlate with telomerase activity and to be implicated in the regulation of telomerase activity in cancer cells[37]. Furthermore, telomerase activity in telomerase-negative cells can be restored by ectopic expression of TERT, suggesting that in certain cases, TERT is the only limiting factor for telomerase activation[38]. The relationship between hypoxia and the induction of telomerase activity should be considered. Hypoxic conditions could induce a DNA damage response by causing telomere damage. In response to this damage, hypoxia-inducible factor-1α may induce telomerase in order to heal the damaged chromosome ends[39].
The cells in the culture underwent senescence. This phenotype was attributable to unsuitable conditions (culture shock, oxidative stress) or to growth arrest mediated by p38 MAPK, and p19Arf, not to telomere shortening per se[40]. hTERT transduction also failed to prevent premature senescence in 21% O2[41]. Maybe the activation of p38 MAPK contributed to the onset of senescence induced by oxidative stress[42]. p38 MAPK contributed to p53 accumulation in Ataxia Telangiectasia/TERT fibroblasts during oxidative stress-induced senescence[43]. p38 MAPK phosphorylated p53 on Ser33 and Ser46 in the NH2 terminal activation domain and thereby regulated the transactivation activity of p53[43,44].
In summary, these data indicate that premature senescence could be induced in neonatal SD rat cardiomyocytes exposed to hypoxia reoxygenation. Although TERT significantly increased, it could not block senescence.
Acknowledgement
We gratefully acknowledge helpful support from Prof Ji-nan ZHANG.
References
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961;25:585-621.
- Von Zglinicki T, Schewe C. Mitochondrial water loss and aging of cells. Cell Biochem Funct 1995;13:181-7.
- Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci USA 1994;91:4130-4.
- Toussaint O, Fuchs SY, Ronai ZA, Isoyama S, Yuko N, Petronilli V, et al. Reciprocal relationships between the resistance to stresses and cellular aging. Ann N Y Acad Sci 1998;851:450-65.
- Saretzki G, Feng J, von Zglinicki T, Villeponteau B. Similar gene expression pattern in senescent and hyperoxic-treated fibroblasts. J Gerontol A Biol Sci Med Sci 1998;53:B438-42.
- Yang NC, Hu ML. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp Gerontol 2005;40:813-9.
- Kang MK, Shin KH, Yip FK, Park NH. Normal human oral keratinocytes demonstrate abnormal DNA end joining activity during premature senescence. Mech Ageing Dev 2005;26:475-9.
- Unterluggauer H, Hampel B, Zwerschke W, Jansen-Durr P. Senescence-associated cell death of human endothelial cells: the role of oxidative stress. Exp Gerontol 2003;38:1149-60.
- Van der Loo B, Fenton MJ, Erusalimsky JD. Cytochemical detection of a senescence-associated beta-galactosidase in endo-thelial and smooth muscle cells from human and rabbit blood vessels. Exp Cell Res 1998;241:309-15.
- Reincke BS, Rosson GB, Oswald BW, Wright CF. INI1 expression induces cell cycle arrest and markers of senescence in malignant rhabdoid tumor cells. J Cell Physiol 2003;194:303-13.
- Meng A, Wang Y, Van Zant G, Zhou D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res 2003;63:5414-9.
- Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res 2003;93:604-13.
- Ball AJ, Levine F. Telomere-independent cellular senescence in human fetal cardiomyocytes. Aging Cell 2005;4:21-30.
- Huang Y, Hickey RP, Yeh JL, Liu D, Dadak A, Young LH, et al. Cardiac myocyte-specific HIF-1 alpha deletion alters vasculariza-tion, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J 2004;18:1138-40.
- Troen BR. The biology of aging. Mt Sinai J Med 2003;70:3-22.
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002;105:1541-4.
- Colgin LM, Reddel RR. Telomere maintenance mechanisms and cellular immortalization. Curr Opin Genet Dev 1999;9:97-103.
- Holzmann B, Nekarda H, Siewert JR. Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J Clin Oncol 2004;22:1807-14.
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998;279:349-52.
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 2000;408:239-47.
- Zhang X, Li J, Sejas DP, Pang Q. Hypoxia-reoxygenation induces premature senescence in FA bone marrow hematopoietic cells. Blood 2005;106:75-85.
- Takeuchi H, Bilchik A, Saha S, Turner R, Wiese D, Tanaka M, et al. c-MET expression level in primary colon cancer: a predictor of tumor invasion and lymph node metastases. Clin Cancer Res 2003;9:1480-8.
- Vasa M, Breitschopf K, Zeiher AM, Dimmeler S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res 2000;87:540-2.
- Von Zglinicki T, Marzabadi MR, Roomans GM. Water and ion distributions in myocytes cultured under oxidative stress mimic changes found in the process of aging. Mech Ageing Dev 1991;58:49-60.
- Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol 1996;40:H2183-9.
- Kawamata N, Inagaki N, Mizumura S, Sugimoto KJ, Sakajiri S, Ohyanagi-Hara M, et al. Methylation status analysis of cell cycle regulatory genes (p16INK4a, p15INK4B, p21Waf1/Cip1, p27Kip1 and p73) in natural killer cell disorders. Eur J Haematol 2005;74:424-9.
- Tevelev A, Byeon IJ, Selby T, Ericson K, Kim HJ, Kraynov V, et al. Tumor suppressor p16INK4a: structural characterization of wild type and mutant proteins by NMR and circular dichroism. Biochemistry 1996;35:9475-87.
- Sherr CJ. Cancer cell cycles. Science 1996;274:1672-7.
- Ausserlechner MJ, Obexer P, Geley S, Kofler R. G1 arrest by p16INK4a uncouples growth from cell cycle progression in leukemia cells with deregulated cyclin E and c-Myc expression. Leukemia 2005;19:1051-7.
- Mouton RE, Venable ME. Ceramide induces expression of the senescence histochemical marker, beta-galactosidase, in human fibroblasts. Mech Ageing Dev 2000;113:169-81.
- Stockschlader MA, Schuening FG, Graham TC, Storb R. Transplantation of retrovirus-transduced canine keratinocytes expressing the beta-galactosidase gene. Gene Ther 1994;1:317-22.
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995;92:9363-7.
- Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol 2001;13:748-53.
- Antropova YG, Bryantsev AL, Kalinina NI. Il抜nskaya OP, Tararak EM. Proliferative activity and expression of cyclin-dependent kinase inhibitor p21WAF1 and p53 protein in endothelial cells of human aorta during replicative aging in vitro. Bull Exp Biol Med 2002;134:81-3.
- Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activa-tion. Mol Cell Biol 2001;21:3336-42.
- Seimiya H, Tanji M, Oh-Hara T, Tomida A, Naasani I, Tsuruo T. Hypoxia up-regulates telomerase activity via mitogen-activated protein kinase signaling in human solid tumor cells. Biochem Biophys Res Commun 1999;260:365-70.
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997;90:785-95.
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998;279:349-52.
- Hirotaka N, Toshihide N, Satoru K, Masaki I, Jerry WS, Keiichi I. Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT). Mol Cell Biol 2004;24:6076-83.
- Naka K, Tachibana A, Ikeda K, Motoyama N. Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J Biol Chem 2004;279:2030-7.
- Martin JA, Klingelhutz AJ, Moussavi-Harami F, Buckwalter JA. Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. J Gerontol A Biol Sci Med Sci 2004;59:324-37.
- Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003;8:131-44.
- Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J 1999;18:6845-54.
- Sanchez-Prieto R, Rojas JM, Taya Y, Gutkind JS. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res 2000;60:2464-72.