
Molecular basis of ataxia telangiectasia and related diseases
Ataxia telangiectasia clinical presentation
Ataxia telangiectasia (AT) is a rare human disease characterized by extreme sensitivity to radiation[1–5]. AT is a progressive neurodegenerative disorder causing a predisposition to cancer, with a hallmark of onset in early childhood[6–9]. AT is seen in approximately 1 in every 40 000 live births in the USA, although the frequency varies from country to country[10]. At birth, infants appear normal and begin walking at a normal age (approximately age 1 year); however, by age 2–3 ataxia (loss of muscle co-ordination) becomes visible and generally by age 10 patients are confined to a wheelchair[10]. Ataxia generally precedes telangiectasia, which is described as the chronic dilation of a group of capillaries causing elevated, dark red blotches on the skin or eyes[11]. This disease is additionally characterized by cerebellar degeneration, and immune system defects[12–14]. In younger children, diagnosis of AT is somewhat obscure as the cerebellum appears to be of normal size for several years although onset of ataxia is prevalent. However, by age 10 magnetic resonance imaging (MRI) generally shows an abnormal cerebellum that has decreased in volume[15]. Studies have shown a gradual decrease in granular and Purkinje cells, which are large branching cells of the nervous system and are located in the middle layer of the lower part of the brain, or the cerebellum[14–16]. Unfortunately, there is currently no treatment for AT except for supportive therapy of secondary symptoms[17].
In addition to an up to several hundredfold increase in developing certain types of cancers (eg lymphoma) in AT patients, epidemiological studies suggest that heterozygote AT mutation carriers are also at increased risk for cancer, particularly breast carcinoma[18,19]. This is of great significance as it is estimated that approximately 1% of the population are AT carriers. However, it is noted that a recent study showed that heterozygous AT mutations did not confer genetic predisposition to early onset of breast cancer[20].
Serum alphafetoprotein (AFP) levels are elevated in more than 95% of AT patients, which is therefore used to diagnose AT and to distinguish from AT variants[4,5,10,21]. When dealing with AFP it is important to note that AFP levels are normally higher at birth than those at age 2, thus diagnostics for AT using AFP cannot be conducted until after the age of 2. There are known malignancies associated with AFP[10,22]. For example, AFP is known to be elevated in case of spina bifida and anencephaly, and this elevation is the hallmark test for early detection of both conditions. Spina bifida is part of a group of birth defects called neural tube defects, which affect embryonic structures that eventually develop into the brain, spinal cord and tissues that enclose them. Anencephaly is a congenital condition in which portions of the brain fail to develop. Because AFP is such a diagnostic hallmark for AT, it is surprising that the mechanism of AFP upregulation in normal and AT cells has not been reported. Nevertheless, given the commonality of AFP elevation in all of the above 3 diseases, it is conceivable that the increased AFP level is responsible for the Purkinje cell degeneration in AT patients.
Ataxia telangiectasia-related diseases
There are several human deficiencies and diseases that are closely related to AT. For example, molecular cloning has now allowed for the distinction between AT and other autosomal recessive cerebellar ataxias (ARCA) such as Friedreich ataxia or A-TFresno, an old-fashioned term for an AT variant[23–25], oculomotor apraxias 1 (aprataxin deficiency), oculomotor apraxias 2 (senataxin deficiency), aicardi syndrome and AT-like disorder (ATLD)[26–29].
AT-like disorder is a very rare disorder with clinical features similar to those of AT, with the most prominent similarity being progressive cerebellar ataxia. However, ATLD patients that do not present with telangiectasia[30,31], have a later onset of neurological features, slower progression, and thus milder symptoms compared to AT patients[27].
Closely related to AT and ATLD is the Nijmegen breakage syndrome (NBS)[23–25]. NBS and ATLD have some overlapping features such as hypersensitivity to ionizing radiation and genome instability, and both are characterized by neurological deficits[32,33]. Thus, NBS was long considered as a clinical variant of AT. Clinical distinction has been made as NBS patients also present with a characteristic facial appearance and microcephaly as well as growth retardation[34]. Clearly the above AT-related human conditions suggest deficiency at common cellular function(s) and genetic pathway(s) with AT.
Laboratory findings in ataxia telangiectasia and ataxia telangiectasia-related patients
Laboratory findings in patients with AT show: immunodeficiencies emerging as decreased IgA, IgE and IgG2 levels[5,21,35–38], characteristic chromosomal aberrations and an increased rate of telomeric shortening[39], radiosensitivity[1–5], as well as sensitivity to other DNA damaging agents and non-DNA damaging agents[40]. Chromosome instability, cell-cycle checkpoint defects[3] and elevated serum levels of AFP[21,22] are prevalent in most AT patient cells. In addition, approximately one-third of AT patients develop cancer, which is usually lymphoid[10].
Both NBS and ATLD cells share a number of cellular phenotypes with AT cells, with the most prominent being the increased sensitivity to ionizing radiation, abnormal cell-cycle checkpoints, chromosome instability, immunodeficiency and accelerated shortening of telomeres[41,42]. In particular, all 3 disorders show an increased level of chromosome translocation in the peripheral blood between the loci of the immunoglobulin and T-cell receptor genes on chromosomes 7 and 14[43,44]. Hence, laboratory findings are most consistent with all 3 diseases belonging to a group of disorders referred to chromosome instability syndromes.
Molecular basis of ataxia telangiectasia and related diseases
AT is the result of mutations in the AT-mutated (ATM) gene, which was discovered in 1995[45,46]. AT patients suffer as a result of over 400 distinct ATM mutations, of which 85% are accounted for by null mutations in the ATM gene[21,47,48]. Thus, approximately 85% of AT sufferers have no detectable ATM protein[21,48]. There are a few reported genuine AT cases with normal ATM protein levels; however, in these cases the protein is defective in ATM enzyme activity[49,50]. The establishment of AT as a monogenetic disease assisted in efficient diagnosis and reevaluation of “AT variants”. The fact that some AT patients carry hypomorphic ATM mutations (ie mutations with partial functions) provides an underlying explanation for atypical AT patients with minimal signs of symptoms, such as very mild or late-onset of syndromes or a subset of disorders[51].
The ATM protein is a member of the phosphatidylinositol 3-kinase-like family of serine/threonine protein kinases (PIKK)[13,52]. It is thus grouped because all PIKK contain a conserved kinase domain initially reported in phosphat- idylinositol 3-kinase. This family represents an atypical subclass of protein kinases responsible for phosphorylation of its substrates on serine or threonine followed by glutamine (SQ or TQ)[53–55]. The mammalian PIKK known to be involved in the DNA damage response are: DNA-PKcs (the catalytic subunit of DNA-dependent protein kinase), ATM (ataxia telangiectasia gene product), ATR (ATM and Rad3-related), mTOR/FRAP (mammalian target of rapamycin/FKBP-rapamycin-associated protein) and ATX/SMG1[13].
The ATM protein is a fairly large (approximately 350 kDa) protein that can be divided into several structural and functional domains (Figure 1A). The first is an FAT domain, which is conserved among the PIKK family of proteins FRAP, ATR and TRRAP; the second is a phosphoinositide 3,4-kinase (PI3K) domain, which ATM has in common with DNA-PKcs; and the third is an FAT carboxy-terminal domain (FATC)[56]. In addition, the amino terminus of the ATM protein contains multiple HEAT (huntingtin, elongation factor 3, A subunit of protein phosphatase 2A and TOR1) repeats[57]. A recent solution structure analysis of the FATC domain[58] revealed an a-helix and a disulfide-bonded loop that undergoes conformational changes. Hence, the FATC domain may regulate protein activity and stability. The precise function of HEAT repeats is currently unknown, although it is speculated to be involved in the interaction with other proteins, as these repeats are anti-parallel α-helices linked by a flexible loop[40,57]. The overall shape of ATM is very similar to DNA-PKcs and is comprised of a head and a long arm that is thought to wrap around double-stranded DNA after a conformational change[59–63]. One of the hallmarks of the ATM protein is its rapid increase in kinase activity immediately following exposure to ionizing radiation (IR), or in the presence of double-strand breaks (DSB)[64,65].
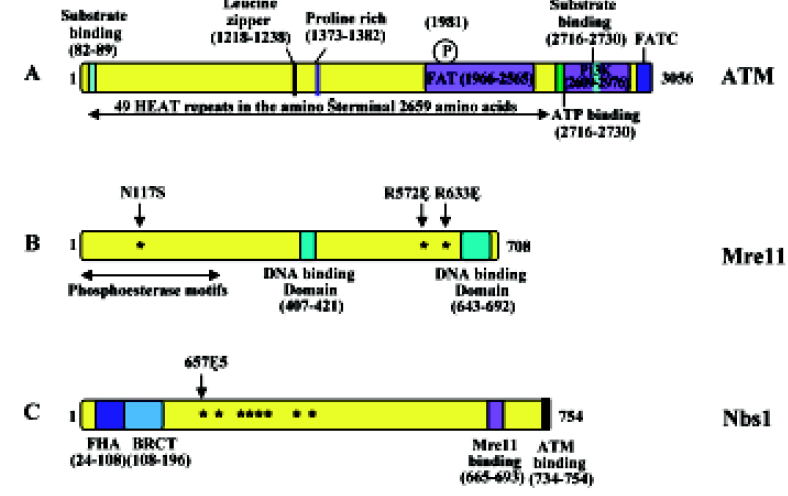
The gene mutated in NBS was identified as NBS1[25,66,67] whereas ATLD is caused by mutations in the MRE11 gene[29]. Unlike AT, which can result from complete inactivation of the coding gene, all NBS and ATLD patients carry hypomorphic mutations that express some level of corresponding protein, either truncated or full length with amino acid substitutions[27,68]. Indeed, attempts to create null Mre11 mutations in mouse embryonic stem cells has failed and conditional knockout experiments demonstrated that this gene is required for normal cell proliferation[69], suggesting that MRE11 is an essential gene in mammals. Similarly, complete Nbs1 knockout mice are embryonic lethal[70,71]. This assertion is further confirmed by a report that disruption of mouse Rad50, encoding the third component of the MRN (MRX in yeast) complex consisting of Mre11, Rad50, and Nbs1 (aka nibrin and p95; Xrs2 in yeast), also causes embryonic stem cell lethality[72].
The human MRE11 gene was initially isolated by its possible protein interaction with DNA ligase I[73] and was predicted to encode a 708-amino acid protein. However, subsequent reports appear to favor an Mre11B sequence (Figure 1B), which is different from Mre11 primarily at the C-terminal region and significantly strengthens its similarity with Mre11 from other species[74]. Mre11 contains a phosphoesterase motif at the N-terminal region that is highly conserved among all eukaryotic Mre11 and a similar motif is also found in Escherichia coli SbcD[75]. It also contains DNA-binding domains at the C-terminal region and region(s) for homodi-merization[76,77]. Structural analyses[78–80] indicate that the MRN complex consists of two molecules of Mre11 and Rad50 (an E coli SbcC homolog) forming a rope-and-hook structure capable of bridging two DSB ends. In vitro characterization of the Rad50–Mre11 complex[76,81–84] demonstrates that it possesses 3´ to 5´ exonuclease and single-strand endonuclease activities, as well as ATP-dependent DNA binding and limited unwinding activity.
The phenotypic similarity between NBS and ATLD patients is attributed to the fact that both corresponding proteins are components of the MRN complex. Mre11 interacts with both Rad50 and Nbs1/Xrs2, and its homodimerization or self-interaction appears to be important for the Mre11–Rad50 and Mre11–Nbs1/Xrs2 interactions[76,77]. Interestingly, deficiency of the Mre11 protein results in decreased cellular levels of Nbs1 and Rad50, suggesting that the MRN complex is required for protein stability. The primary cellular function of MRN is a sensor for DNA strand breaks and to activate signaling pathways leading to cell-cycle checkpoint and recombination repair[32,85,86]. While Mre11 and Rad50 are highly conserved in eukaryotes, from unicellular yeasts to human cells[87,88], the third component, Nbs1, is less conserved structurally among eukaryotes, although the mammalian Nbs1 and yeast Xrs2 play similar roles within their respective MRN/MRX complex. Nbs1 has been suggested to be a regulatory subunit of MRN that is essential for Mre11 phosphorylation upon DNA damage[89] and its biochemical activities, such as ATP-dependent DNA unwinding and nuclease activity[90]. Although Nbs1 or Xrs2 is not required for the Mre11–Rad50 enzymatic activity in vitro, its activity appears to be absolutely required for MRX activity in vivo, as inactivation of any 1 of the 3 genes in yeast cells leads to complete loss of the MRX activity and indistinguishable cellular phenotypes[91–93]. This essential function of Nbs1/Xrs2 is probably due to its role in recruiting ATM/Tel1[94]. The Nbs1 protein has 4 known functional regions: the N-terminal forkhead-associated (FHA) domain followed by a BRCA1 C-terminus (BRCT) domain, a C-terminal Mre11-binding domain and an ATM-binding domain at the extreme C-terminus (Figure 1C). Interestingly, over 90% of all NBS patients analyzed to date contain a homozygous 5 bp truncating mutation, 657Δ5[25], resulting in the production of a 26-kDa protein with FHA and BRCT domains but lacking the Mre11-binding domain (Figure 1C). Knockout mice producing similar truncated Nbs1 proteins are viable and develop symptoms characteristic of the human disease[95,96], thus providing an animal model suitable for NBS research.
Phenotypic manifestation of genetic defects in ataxia telangiectasia and related diseases
One of the hallmarks of the ATM protein is its rapid increase in kinase activity immediately following DSB formation[64,65], and its kinase activity remains its only known function to date. The phenotypic manifestation of AT is due to the broad range of substrates for the ATM kinase, as compiled recently[40], involving DNA repair, apoptosis, G1/S, intra-S checkpoint and G2/M checkpoints, gene regulation, translation initiation, and telomere maintenance. Protein phosphorylation and de-phosphorylation is an important cellular regulatory mechanism that governs protein activity, stability, subcellular location and complex formation. We propose that although the primary ATM kinase substrates appear to be related to cellular response to DNA damage, novel ATM substrates and subsequent effects may explain all observed syndromes of AT patients, including elevated AFP level. We also propose that the multiple HEAT repeats in ATM may serve as a platform to interact with various regulatory and substrate proteins and to control its kinase activity in the aftermath of DNA damage. For example, the increased risk for breast cancer in AT patients has been implicated by the involvement of ATM in the interaction and phosphorylation of BRCA1 and its associated proteins following DNA damage[97,98].
The ATM protein also has the ability to interact with the ends of double-stranded DNA[99]. A recent model suggests that ATM is in an inactive dimeric or polymeric configuration, in which the kinase domain of each molecule is blocked by the FAT domain of the other[100]. Following DNA damage, each ATM molecule phosphorylates the other on a serine residue at position 1981 within the FAT domain and thus converts into fully active monomers. It was shown that protein serine-threonine phosphatase 5 (PP5) is required for the activation of ATM and its subsequent kinase activity[101].
The symptom similarity among AT, ATLD and NBS is apparently due to physical and genetic interactions between ATM and MRN. Experimental results suggest that the ATM-MRN interaction in response to IR is complicated. First, the histone H2A isoform H2AX is rapidly phosphorylated (within 5 min) after irradiation in an ATM-dependent manner[102,103], and the phosphorylated gH2AX interacts with MRN via the FHA/BRCT region of Nbs1[103]. This interaction has been deemed indispensable for the recruitment of MRN[41]. Second, in response to IR, ATM phosphorylates Nbs1 in vivo[104], which is required for the subsequent phosphorylation of Mre11. Finally, phosphorylation and activation of the S-phase checkpoint effector Smc1 by ATM requires phosphorylated Nbs1[105,106]. The above observations suggest that ATM acts upstream of MRN and is required to recruit MRN to the damage sites observed as distinct nuclear foci. On the other hand, MRN/MRX is known to bind DSB ends and a careful choreographic analysis in yeast[107] indicates that MRX localization to nuclear DSB foci precedes and is required for that of Tel1, a yeast ATM ortholog[108,109]. In mammalian cells, Mre11 appears to be required for ATM activation as well[110], and this interaction was further reinforced very recently by an in vitro experiment showing that the MRN complex acts as a DSB sensor for ATM and recruits ATM to broken DNA molecules[111]. Interestingly, the unwinding of DNA ends by MRN appears to be essential for ATM stimulation[111], indicating that the MRN enzymatic activity plays a role in this process. In addition, a recent study[112] shows that NFBD/MDC1, which interacts with MRN, is required for ATM activation under certain conditions. Alternatively, NFBD1/MDC1 may help to recruit substrates, including Nbs1[113,114], to ATM[113–123]; suppression of NFBD1/MDC1 leads to decreased ATM activation and decreased phosphorylation of ATM substrates[112]. These observations support an assertion that MRN plays a crucial role in ATM phosphorylation of other substrates in response to DSB. The argument that MRN functions upstream of ATM is further strengthened by a very recent report that the extreme C-terminal 21 amino acid peptide of Nbs1 is required and sufficient for the interaction with ATM in vivo and in vitro, and that this interaction mediates ATM deposition to the sites of DNA damage and its checkpoint functions[94]. The above genetic and physical interactions between MRN and ATM underlie the molecular basis for the shared cellular functions in cell-cycle checkpoints and recombination repair of DSB, as well as common syndromes among AT, ATLD and NBS.
DNA-PK: a missing link?
Despite the great advances made in the past decade regarding AT research, many questions remain to be answered. One question that remains unsolved is the involvement of Ku70/Ku80 in ATM-mediated cellular events in response to DSB. Ku70 and Ku80 form a heterodimer as DNA end-binding subunits of DNA-PK[59–62]. It would be plausible to suggest that Ku70/Ku80 may also assist ATM’s translocation to DSB, as Ku70/Ku80 is known to play such a role for DNA-PKcs, another member of PIKK with overall structural similarity to ATM[124]. Like ATM, DNA-PKcs recognizes the same consensus phosphorylation sequence SQ/TQ, and also undergoes DSB-induced autophosphorylation, which is required for DSB repair[125]. In addition, inactivation of either DNA-PK or ATM results in immunodeficiency; the former results in defective V(D)J recombination that leads to scid (severe combined immune deficiency) mutations[126,127], whereas the latter results in the disruption of immune gene rearrangement[128]. We propose that in the presence of Ku70/Ku80, DNA-PKcs and ATM may undergo conformational changes to be suitable for the suggested role of further wrapping around DNA[59–63]. Indeed, recent electron microscopy-based structural analyses of both DNA-PKcs[129–131] and ATM[63] suggest a similar architecture consisting of a “head” and a “palm” domain connected by an “arm”. The palm domain binds DNA, which induces conformational changes and triggers an interaction between initially distant palm and head regions[130]. These DNA-induced conformational changes alter the catalytic core and regulate the kinase activity[131]. The difference between DNA-PKcs and ATM is that DNA-PKcs is activated in G1 and is required for non-homologous end joining (NHEJ) and apoptosis if DNA damage is excessive[132], while ATM is activated in S and G2 phases and is required for S-phase checkpoint and homologous recombination (HR)[40].
Another connection between ATM and Ku70/Ku80 is that yeast MRX is not only required for DSB signaling and HR, but also works with Ku70/Ku80 for NHEJ[133–136] and telomere maintenance[77,93,137,138]. The budding yeast Saccarom- yces cerevisiae predominantly employs HR to repair DSB over NHEJ. On some occasions when DSB repair can be processed by either process, NHEJ was shown to precede HR[139]. A compiled model can be proposed in which the telomere acts as a repository for Ku proteins, which relocalize to DSB after DNA damage[140] and recruit MRX[141]. MRX can assist to recruit and activate Lif1 and Dnl4[142], resulting in ligation of the broken ends. However, in budding yeast, the MRX complex most likely initiates 5´ to 3´ end resection[76], hence committing to repair by HR. This commitment is probably due to the broken DSB ends that are unsuitable for direct ligation, or to the recruitment of Tel1 by MRX that activates cell-cycle checkpoints and subsequent recruitment of other HR proteins[107]. A similar regulatory cascade may also exist in higher eukaryotes; however, because mammalian cells have an extended G1 phase, the initial DSB binding by Ku70/Ku80 may stimulate NHEJ more frequently than in yeast. In addition, mammalian cells contain 3 major PIKK family proteins, namely ATM, ATR and DNA-PKcs, involved in different modes of DNA repair and overlapping signal transduction in response to DNA damage. The mechanism of their recruitment to sites of DNA damage is conserved via a newly discovered motif[94]. For example, ATR is recruited to a single-stranded DNA by ATRIP (ATR-interacting protein)[143,144], whereas DNA-PKcs and ATM are recruited to DSB by Ku70/K80 and MRN, respectively. Based on the above observations, we have proposed a working model of cellular response to DSB in mammals, as presented in Figure 2.
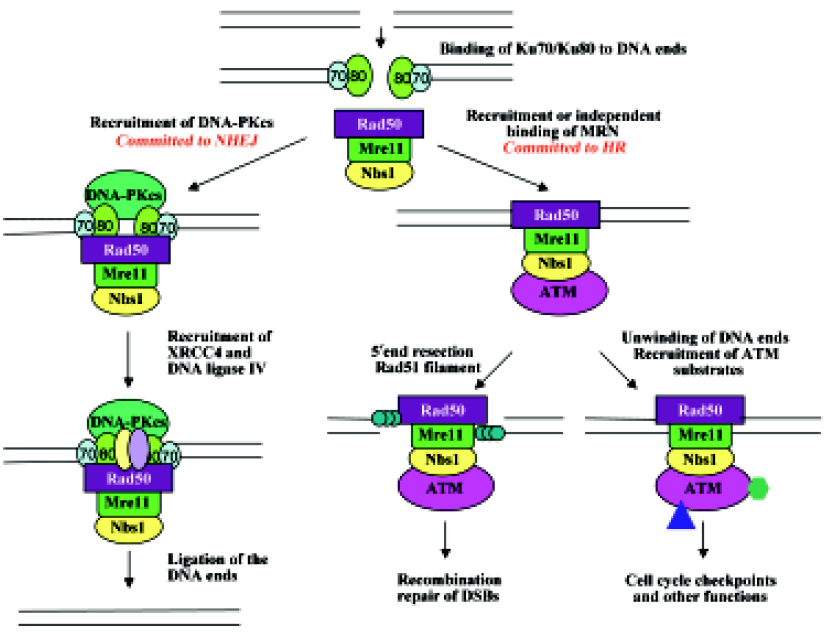
Finally, the physical and functional interactions among Ku, MRN/MRX and ATM/Tel1 can also be found in telomeres. Both yeast[145] and human[146] Ku is bound to telomeric DNA and protects its 3´ overhang. The MRX complex in budding yeast is also associated with telomeres, and mutants deficient in Ku or MRX activity exhibit telomere shortening[137,147]. Similar phenotypes are also observed in their homologous mutants in fission yeast and plants[148–150]. Tel1 works together with MRX by either regulating the access of telomerase to the DNA end[151], and/or preventing telomere end-to-end fusion[152]. Tel1 is activated by the MRX complex and in turn phosphorylates MRX[153], suggesting functional conservation between yeast and mammals. However, unlike Tel1, ATM has not been implicated in telomerase access to the telomere. Nevertheless, AT patient cells were shown to have shortened telomeres[39,154], as well as other telomere abnormalities[155,156]. Furthermore, transgenic mice defective in both ATM and telomerase dramatically increase telomere dysfunction compared to single mutants[157], suggesting roles of ATM in telomere maintenance. The telomeric functions of ATM may be related to some symptoms of AT patients.
Conclusions
Since the initial discovery of the AT mutated gene 10 years ago, knowledge regarding the molecular mechanisms of AT and its related diseases has accumulated rapidly. It is now clear that the primary function of ATM and MRN, which is related to NBS and ATLD, is to respond to DNA damage, especially DSB. Two primary activities are cell-cycle checkpoint and DNA repair through HR, and these are achieved mainly via protein phosphorylation of various substrates, including ATM and MRN themselves. ATM and MRN are also involved in telomere maintenance and protection from chromosomal translocation, as well as efficient immune gene recombination. Although these cellular phenotypes are able to explain most phenotypes observed in AT and AT-related disease, some other associated symptoms may be due to diverse ATM substrates, unique MRN functions not shared by ATM, as well as the hypomorphic nature of the disease. Despite great advances in research, many questions remain to be answered especially with regard to treatment. Future efforts shall be directed to translational research for the cure and prevention of AT its and related diseases.
Acknowledgements
The authors wish to thank Michelle HANNA for proofreading the manuscript and other laboratory members for their helpful discussion. This work is supported by a Canadian Institutes of Health Research operating grant (MOP-53240) to WX.
References
- Gotoff SP, Amirmokri E, Liebner EJ. Ataxia telangiectasia. Neoplasia, untoward response to x-irradiation, and tuberous sclerosis. Am J Dis Child 1967;114:617-25.
- Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, et al. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature 1975;258:427-29.
- Young BR, Painter RB. Radioresistant DNA synthesis and human genetic diseases. Hum Genet 1989;82:113-7.
- Huo YK, Wang Z, Hong JH, Chessa L, McBride WH, Perlman SL, et al. Radiosensitivity of ataxia-telangiectasia, X-linked agamm- aglobulinemia, and related syndromes using a modified colony survival assay. Cancer Res 1994;54:2544-7.
- Sun X, Becker-Catania SG, Chun HH, Hwang MJ, Huo Y, Wang Z, et al. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J Pediatr 2002;140:724-31.
- Boder E, Sedgwick RP. Ataxia-telangiectasia: a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics 1958;21:526-54.
- Boder E. Ataxia-telangiectasia: an overview. Kroc Found Ser 1985;19:1-63.
- Gatti RA, Boder E, Vinters HV, Sparkes RS, Norman A, Lange K. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine (Baltimore) 1991;70:99-117.
- Gatti RA. Ataxia-telangiectasia. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. New York: Mc Graw-Hill; 2002. p 239–65.
- Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 2004;3:1187-96.
- Cabana MD, Crawford TO, Winkelstein JA, Christensen JR, Lederman HM. Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics 1998;102:98-100.
- Shiloh Y. ATM: ready, set, go. Cell Cycle 2003;2:116-7.
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003;3:155-68.
- Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol 1997;15:177-202.
- Tavani F, Zimmerman RA, Berry GT, Sullivan K, Gatti R, Bingham P. Ataxia-telangiectasia: the pattern of cerebellar atrophy on MRI. Neuroradiology 2003;45:315-9.
- Nowak R. Discovery of AT gene sparks biomedical research bonanza. Science 1995;268:1700-1.
- Perlman S, Becker-Catania S, Gatti RA. Ataxia-telangiectasia: diagnosis and treatment. Semin Pediatr Neurol 2003;10:173-82.
- Swift M, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med 1991;325:1831-6.
- Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med 1987;316:1289-94.
- FitzGerald MG, Bean JM, Hegde SR, Unsal H, MacDonald DJ, Harkin DP, et al. Heterozygous ATM mutations do not contribute to early onset of breast cancer. Nat Genet 1997;15:307-10.
- Chun HH, Sun X, Nahas SA, Teraoka S, Lai CH, Concannon P, et al. Improved diagnostic testing for ataxia-telangiectasia by immunoblotting of nuclear lysates for ATM protein expression. Mol Genet Metab 2003;80:437-43.
- McVey JH, Michaelides K, Hansen LP, Ferguson-Smith M, Tilghman S, Krumlauf R, et al. A G→A substitution in an HNF I binding site in the human alpha-fetoprotein gene is associated with hereditary persistence of alpha-fetoprotein (HPAFP). Hum Mol Genet 1993;2:379-84.
- Weemaes CM, Hustinx TW, Scheres JM, van Munster PJ, Bakkeren JA, Taalman RD. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr Scand 1981;70:557-64.
- Heil JA, Weemaes CM, van den Heuvel LP, van Engelen BG, Gabreels FJ, Smeets DF, et al. International Nijmegen Breakage Syndrome Study Group. Nijmegen breakage syndrome. Arch Dis Child 2000;82:400-406.
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998;93:467-76.
- Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 2004;36:225-7.
- Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD) – its clinical presentation and molecular basis. DNA Repair (Amst) 2004;3:1219-25.
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 2001;29:184-8.
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999;99:577-87.
- Klein C, Wenning GK, Quinn NP, Marsden CD. Ataxia without telangiectasia masquerading as benign hereditary chorea. Mov Disord 1996;11:217-20.
- Hernandez D, McConville CM, Stacey M, Woods CG, Brown MM, Shutt P, et al. A family showing no evidence of linkage between the ataxia telangiectasia gene and chromosome 11q22-23. J Med Genet 1993;30:135-40.
- D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002;3:317-27.
- Petrini JH. The Mre11 complex and ATM: collaborating to navigate S phase. Curr Opin Cell Biol 2000;12:293-6.
- Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002;420:93-8.
- Sanal O, Ersoy F, Yel L, Tezcan I, Metin A, Ozyurek H, et al. Impaired IgG antibody production to pneumococcal polysaccharides in patients with ataxia-telangiectasia. J Clin Immunol 1999;19:326-34.
- Gatti RA. The inherited basis of human radiosensitivity. Acta Oncol 2001;40:702-11.
- Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol 1999;9:699-702.
- Bakhshi S, Cerosaletti KM, Concannon P, Bawle EV, Fontanesi J, Gatti RA, et al. Medulloblastoma with adverse reaction to radiation therapy in Nijmegen breakage syndrome. J Pediatr Hematol Oncol 2003;25:248-51.
- Metcalfe JA, Parkhill J, Campbell L, Stacey M, Biggs P, Byrd PJ, et al. Accelerated telomere shortening in ataxia telangiectasia. Nat Genet 1996;13:350-3.
- Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889-900.
- Kobayashi J, Antoccia A, Tauchi H, Matsuura S, Komatsu K. NBS1 and its functional role in the DNA damage response. DNA Repair (Amst) 2004;3:855-61.
- Tauchi H, Matsuura S, Kobayashi J, Sakamoto S, Komatsu K. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene 2002;21:8967-80.
- Chrzanowska KH, Kleijer WJ, Krajewska-Walasek M, Bialecka M, Gutkowska A, Goryluk-Kozakiewicz B, et al. Eleven Polish patients with microcephaly, immunodeficiency, and chromosomal instability: the Nijmegen breakage syndrome. Am J Med Genet 1995;57:462-71.
- van der Burgt I, Chrzanowska KH, Smeets D, Weemaes C. Nijmegen breakage syndrome. J Med Genet 1996;33:153-6.
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995;268:1749-53.
- Savitsky K, Sfez S, Tagle DA, Ziv Y, Sartiel A, Collins FS, et al. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum Mol Genet 1995;4:2025-32.
- Gilad S, Chessa L, Khosravi R, Russell P, Galanty Y, Piane M, et al. Genotype–phenotype relationships in ataxia-telangiectasia and variants. Am J Hum Genet 1998;62:551-61.
- Becker-Catania SG, Chen G, Hwang MJ, Wang Z, Sun X, Sanal O, et al. Ataxia-telangiectasia: phenotype/genotype studies of ATM protein expression, mutations, and radiosensitivity. Mol Genet Metab 2000;70:122-33.
- Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, et al. ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet 1998;62:334-45.
- Stewart GS, Last JI, Stankovic T, Haites N, Kidd AM, Byrd PJ, Residual ataxia telangiectasia mutated protein function in cells from ataxia telangiectasia patients, with 5762ins137 and 7271T–>G mutations, showing a less severe phenotype. J Biol Chem 2001; 276: 30 133–41.
- Lavin MF, Scott S, Gueven N, Kozlov S, Peng C, Chen P. Functional consequences of sequence alterations in the ATM gene. DNA Repair (Amst) 2004;3:1197-205.
- Goodarzi AA, Block WD, Lees-Miller SP. The role of ATM and ATR in DNA damage-induced cell cycle control. Prog Cell Cycle Res 2003;5:393-411.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002;298:1912-34.
- Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 1999; 274: 37 538–43.
- O’Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, Abraham RH, . Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem 2000; 275: 22 719–27.
- Bosotti R, Isacchi A, Sonnhammer EL. FAT: a novel domain in PIK-related kinases. Trends Biochem Sci 2000;25:225-7.
- Perry J, Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 2003;112:151-5.
- Dames SA, Mulet JM, Rathgeb-Szabo K, Hall MN, Grzesiek S. The solution structure of the FATC domain of the protein kinase TOR suggests a role for redox-dependent structural and cellular stability. J Biol Chem 2005;280:20558-64.
- Cary RB, Peterson SR, Wang J, Bear DG, Bradbury EM, Chen DJ. DNA looping by Ku and the DNA-dependent protein kinase. Proc Natl Acad Sci USA 1997;94:4267-72.
- Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev 1999;13:916-34.
- Merkle D, Douglas P, Moorhead GB, Leonenko Z, Yu Y, Cramb D, . The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry 2002; 41: 12 706–14.
- Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J 1997;16:5098-112.
- Llorca O, Rivera-Calzada A, Grantham J, Willison KR. Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an ATM-like domain to clamp around double-stranded DNA. Oncogene 2003;22:3867-74.
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998;281:1677-9.
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998;281:1674-7.
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR 3rd, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 1998;93:477-86.
- Matsuura S, Tauchi H, Nakamura A, Kondo N, Sakamoto S, Endo S, et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet 1998;19:179-81.
- Digweed M, Sperling K. Nijmegen breakage syndrome: clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:1207-17.
- Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res 1997;25:2985-91.
- Dumon-Jones V, Frappart PO, Tong WM, Sajithlal G, Hulla W, Schmid G, et al. Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res 2003;63:7263-9.
- Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol 2001;11:105-9.
- Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA 1999;96:7376-81.
- Petrini JH, Walsh ME, DiMare C, Chen XN, Korenberg JR, Weaver DT. Isolation and characterization of the human MRE11 homologue. Genomics 1995;29:80-6.
- Chamankhah M, Wei YF, Xiao W. Isolation of hMRE11B: failure to complement yeast mre11 defects due to species-specific protein interactions. Gene 1998;225:107-16.
- Sharples GJ, Leach DR. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol Microbiol 1995;17:1215-7.
- Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T. Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell 1998;95:705-16.
- Chamankhah M, Xiao W. Formation of the yeast Mre11-Rad50-Xrs2 complex is correlated with DNA repair and telomere maintenance. Nucleic Acids Res 1999;27:2072-9.
- Anderson DE, Trujillo KM, Sung P, Erickson HP. Structure of the Rad50 x Mre11 DNA repair complex from by electron microscopy. J Biol Chem 2001; 276: 37 027–33.
- Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 2001;105:473-85.
- de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell 2001;8:1129-35.
- Trujillo KM, Yuan SS, Lee EY, Sung P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J Biol Chem 1998;273:21447-50.
- Paull TT, Gellert M. The 3' to 5' exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol Cell 1998;1:969-79.
- Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev 1999;13:1276-88.
- Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol 1999;19:556-66.
- Haber JE. The many interfaces of Mre11. Cell 1998;95:583-6.
- Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 2002;66:630-70.
- Anderson RA, Boronenkov IV, Doughman SD, Kunz J, Loijens JC. Phosphatidylinositol phosphate kinases, a multifaceted family of signaling enzymes. J Biol Chem 1999;274:9907-10.
- Kunz J, Wilson MP, Kisseleva M, Hurley JH, Majerus PW, Anderson RA. The activation loop of phosphatidylinositol phosphate kinases determines signaling specificity. Mol Cell 2000;5:1-11.
- Dong Z, Zhong Q, Chen PL. The Nijmegen breakage syndrome protein is essential for Mre11 phosphorylation upon DNA damage. J Biol Chem 1999;274:19513-6.
- Steimle PA, Hoffert JD, Adey NB, Craig SW. Polyphosphoinositides inhibit the interaction of vinculin with actin filaments. J Biol Chem 1999;274:18414-20.
- Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol 1994;14:3414-25.
- Johzuka K, Ogawa H. Interaction of Mre11 and Rad50: two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics 1995;139:1521-32.
- Chamankhah M, Fontanie T, Xiao W. The Saccharomyces cerevisiae mre11(ts) allele confers a separation of DNA repair and telomere maintenance functions. Genetics 2000;155:569-76.
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005;434:605-11.
- Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of Nijmegen breakage syndrome. Curr Biol 2002;12:648-53.
- Kang J, Bronson RT, Xu Y. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J 2002;21:1447-55.
- Chen J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res 2000;60:5037-9.
- Li S, Ting NS, Zheng L, Chen PL, Ziv Y, Shiloh Y, et al. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000;406:210-5.
- Smith GC, Cary RB, Lakin ND, Hann BC, Teo SH, Chen DJ, Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci USA 1999; 96: 11 134–9.
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003;421:499-506.
- Ali A, Zhang J, Bao S, Liu I, Otterness D, Dean NM, et al. Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev 2004;18:249-54.
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999;146:905-16.
- Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, et al. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 2002;12:1846-51.
- Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature 2000;405:473-7.
- Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in ATM-dependent and independent responses to DNA damage. Genes Dev 2002;16:560-70.
- Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 2002;16:571-82.
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 2004;118:699-713.
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell 1995;82:823-9.
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 1995;82:831-40.
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003;22:5612-21.
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005;308:551-4.
- Mochan TA, Venere M, DiTullio RA Jr, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res 2003;63:8586-91.
- Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 2003;421:952-6.
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003;421:961-6.
- Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science 2002;298:1435-8.
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature 2003;421:957-61.
- Shang YL, Bodero AJ, Chen PL. NFBD1, a novel nuclear protein with signature motifs of FHA and BRCT, and an internal 41-amino acid repeat sequence, is an early participant in DNA damage response. J Biol Chem 2003;278:6323-9.
- Xu X, Stern DF. NFBD1/KIAA0170 is a chromatin-associated protein involved in DNA damage signaling pathways. J Biol Chem 2003;278:8795-803.
- Peng A, Chen PL. NFBD1, like 53BP1, is an early and redundant transducer mediating Chk2 phosphorylation in response to DNA damage. J Biol Chem 2003;278:8873-6.
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 2002;4:993-7.
- DiTullio RA Jr, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol 2002;4:998-1002.
- Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol 2003;23:2556-63.
- Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, . Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J Biol Chem 2003; 278: 14 971–7.
- Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, et al. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 1995;82:849-56.
- Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 2002;16:2333-8.
- Hendrickson EA, Qin XQ, Bump EA, Schatz DG, Oettinger M, Weaver DT. A link between double-strand break-related repair and V(D)J recombination: the scid mutation. Proc Natl Acad Sci USA 1991;88:4061-5.
- Blunt T, Finnie NJ, Taccioli GE, Smith GC, Demengeot J, Gottlieb TM, et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 1995;80:813-23.
- Cohen MM, Levy HP. Chromosome instability syndromes. Adv Hum Genet 1989; 18: 43–149, 365-171.
- Leuther KK, Hammarsten O, Kornberg RD, Chu G. Structure of DNA-dependent protein kinase: implications for its regulation by DNA. EMBO J 1999;18:1114-23.
- Boskovic J, Rivera-Calzada A, Maman JD, Chacon P, Willison KR, Pearl LH, et al. Visualization of DNA-induced conformational changes in the DNA repair kinase DNA-PKcs. EMBO J 2003;22:5875-82.
- Rivera-Calzada A, Maman JP, Spagnolo L, Pearl LH, Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure (Camb) 2005;13:243-55.
- Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:909-18.
- Yu J, Marshall K, Yamaguchi M, Haber JE, Weil CF. Microhomology-dependent end joining and repair of transposon-induced DNA hairpins by host factors in Saccharomyces cerevisiae. Mol Cell Biol 2004;24:1351-64.
- Kramer KM, Brock JA, Bloom K, Moore JK, Haber JE. Two different types of double-strand breaks in Saccharomyces cerevisiae are repaired by similar RAD52-independent, nonhomologous recombination events. Mol Cell Biol 1994;14:1293-301.
- Lewis LK, Resnick MA. Tying up loose ends: nonhomologous end-joining in Saccharomyces cerevisiae. Mutat Res 2000;451:71-89.
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol 1996;16:2164-73.
- Boulton SJ, Jackson SP. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J 1998;17:1819-28.
- Nugent CI, Bosco G, Ross LO, Evans SK, Salinger AP, Moore JK, et al. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol 1998;8:657-60.
- Frank-Vaillant M, Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell 2002;10:1189-99.
- Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 1999;97:621-33.
- Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell 2001;8:1105-15.
- Teo SH, Jackson SP. Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr Biol 2000;10:165-8.
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science 2001;294:1713-6.
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003;300:1542-8.
- Gravel S, Larrivee M, Labrecque P, Wellinger RJ. Yeast Ku as a regulator of chromosomal DNA end structure. Science 1998;280:741-4.
- Hsu HL, Gilley D, Blackburn EH, Chen DJ. Ku is associated with the telomere in mammals. Proc Natl Acad Sci USA 1999; 96: 12 454–8.
- Kironmai KM, Muniyappa K. Alteration of telomeric sequences and senescence caused by mutations in RAD50 of Saccharomyces cerevisiae. Genes Cells 1997;2:443-55.
- Gallego ME, White CI. RAD50 function is essential for telomere maintenance in Arabidopsis. Proc Natl Acad Sci USA 2001;98:1711-6.
- Wilson S, Warr N, Taylor DL, Watts FZ. The role of Schizosacc- haromyces pombe Rad32, the Mre11 homologue, and other DNA damage response proteins in non-homologous end joining and telomere length maintenance. Nucleic Acids Res 1999;27:2655-61.
- Ueno M, Nakazaki T, Akamatsu Y, Watanabe K, Tomita K, Lindsay HD, et al. Molecular characterization of the Schizosacc- haromyces pombe nbs1+ gene involved in DNA repair and telomere maintenance. Mol Cell Biol 2003;23:6553-63.
- Ritchie KB, Petes TD. The Mre11p/Rad50p/Xrs2p complex and the Tel1p function in a single pathway for telomere maintenance in yeast. Genetics 2000;155:475-9.
- Chan SW, Blackburn EH. Telomerase and ATM/Tel1p protect telomeres from nonhomologous end joining. Mol Cell 2003;11:1379-87.
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 2001;7:1255-66.
- Xia SJ, Shammas MA, Shmookler Reis RJ. Reduced telomere length in ataxia-telangiectasia fibroblasts. Mutat Res 1996;364:1-11.
- Hande MP, Balajee AS, Tchirkov A, Wynshaw-Boris A, Lansdorp PM. Extra-chromosomal telomeric DNA in cells from Atm(-/-) mice and patients with ataxia-telangiectasia. Hum Mol Genet 2001;10:519-28.
- Pandita TK, Dhar S. Influence of ATM function on interactions between telomeres and nuclear matrix. Radiat Res 2000;154:133-9.
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, et al. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 2003;421:643-8.