
Peroxisome proliferator-activated receptor γ ligands induce cell cycle arrest and apoptosis in human renal carcinoma cell lines1
Introduction
Peroxisome proliferator-activated receptor γ (PPARγ) belongs to the superfamily of nuclear hormone receptor transcription factors[1–3]. PPARγ forms a heterodimer with another nuclear receptor, retinoid X receptor α (RXRα)[4]. In the presence of their ligands, PPARγ/RXRα heterodimer binds to a specific DNA sequence designated peroxisome proliferating response element (PPRE) located in the promoter region of PPARγ target genes and modulates their transcription[5]. Thus far, hundreds of PPARγ target genes have been identified and most of them are involved in adipogenesis[6], glucose metabolism[7] and angiogenesis[8]. Since the discovery of specific ligands for PPARγ, including synthetic antidiabetic thiazolidinediones (TZD) and endogenous 15-deoxy-D12,14-PGJ2 (15dPGJ2) and some polyunsaturated fatty acids, accumulating evidence using many experimental systems suggests that PPARγ plays an important role in carcinogenesis in both adipose and nonadipose cells. It has been known that PPARγ expression is up-regulated in many tumor tissues and its ligands, including pioglitazone, troglitazone and roglitazone, can induce apoptosis and exert antiproliferative effects in human colon cancer[9], breast cancer[10], pituitary adenomas[11], gastric cancer[12] and bladder cancer[13]. Moreover, PPARγ may play an important role in inducing cell differentiation or growth inhibition in patients with liposarcoma[14] and prostate cancer[15]. Therefore activation of PPARγ seems to be a potential approach for treatment of some malignant tumors.
Renal cell carcinoma (RCC) is the most common renal tumor and the third malignancy within urological oncology. It makes up approximately 2%–3% of all adult malignancies. At present, more than 50% of all RCC are found incidentally, which results in a high proportion of patients with progressive metastasis at the time of diagnosis[16,17]. Surgery remains the only effective therapeutic option. Although combination of chemotherapy and/or radiotherapy might help in survival rate, it is considered to be of limited value for the treatment of RCC. Therefore, improvement of life span in patients with renal cell carcinoma greatly depends on the identification of novel treatment strategies. An alternative treatment may include activation of PPARγ, as PPARγ has been shown to be expressed in the kidney[18], and its ligands can induce cell growth inhibition and terminal differentiation in many other maliganancies[9–14].
In the present study, we examined the expression of PPARγ in human primary RCC and RCC cell lines, and determined the biological events of PPARγ activation in inducing RCC cell cycle arrest and apoptosis. We have also explored the molecular mechanism through which PPARγ agonists inhibit cell growth and induce cell death in RCC cell lines.
Materials and methods
Chemicals Pioglitazone and troglitazone were kindly gifted by Park-Davis Pharmaceutical Research (Ann Arbor, MI, USA) and Takeda Chemical Industries (Osaka, Japan). All other chemicals were purchased from Sigma (St Louis, MO, USA). Stock solutions of pioglitazone or troglitazone were made at 100 mmol/L concentration in dimethyl sulfoxide and added to the culture medium at the final concentration of less than 0.1%.
Cell lines and culture conditions Human RCC-derived cell lines 786-O and A498, human proximal tubular cell line (HK-2), and human mesangium cell line (HMCL) were cultured in Hepes-buffered RPMI-1640 medium (Gibco BRL, Grand Island, NY, USA) supplemented with 10% FBS, 100 kU/mL penicillin G, 100 g/L streptomycin, and 2 mmol/L l-glutamine in a humidified 5% CO2 atmosphere at 37 ºC. The media were changed every 3 d, and the cells were separated by trypsinization using trypsin/edetic acid when they reached 90% confluence.
Tumor specimens and immunohistochemistry Specimens were obtained from 120 patients with RCC and 20 patients with normal kidney (NK) who underwent total nephroureterectomy as a result of ureteral cancer or trauma. All patients were treated at the Institute of Urology in Peking University between June 1999 and June 2001. No patients had received irriadiation or chemotherapy prior to surgery. Tumor samples with the highest nuclear grade were selected. All tissues were preserved in 10% formalin serially sectioned onto microscope slides with a thickness of 4 µmol/L. Sections were immunostained with polycolonal antibody to human PPARg (1:100, Santa Cruz Biotechonology, Santa Cruz, CA, USA) using avidin-biotin-complex-peroxidase and counterstained with hematoxylin. Negative control slides were prepared by omitting the primary antibody. The intensity of immunostaining and the ratio of the positive cells were roughly graded into four scores (0, 1, 2, 3, and 4) by two observers who did not know the origin of the samples on two occasions. The score 4 was defined as maximum intensity of immunostaining throughout the section, while the score 0 implied that staining was absent throughout the specimen.
Measurement of PPARγ mRNA by reverse transcription-polymerase chain reaction Total RNA from 15 RCC tissues, 10 NK tissues, RCC cell lines 786-O and A498, HK-2 cells and HMCC (normal kidney derived mesangium cell line). Cells was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Contaminated genomic DNA in total RNA was removed by RNase-free DNase I digestion. Reverse transcription was performed in a total volume of 25 µL using 3 µg of total RNA, oligo-dT primer and MMLV reverse transcriptase (Amersham, Buckinghamshire, UK). Primers used for the amplification of PPARγ cDNA were synthesized at the SBS Gene Company (Beijing, China). The sense primer was 5'-AGAGATGCCATTCTGGCC-3' and antisense primer was 5'-GTGGAGTAGAAATGCTGGAGA-3'. Polymerase chain reaction (PCR) amplification yielded a PCR product of 130 bp. Primers used for amplifying a 500-bp of β-actin cDNA as an internal control were as follows: 5'-ACTGACTACCTCA-TGAAGATC-3' (sense) and 5'-CGTCATACTCCTGCTTGC-3' (anti-sense). The PCR condition was 40 s (denaturation) at 94 °C, 35 s (annealing) at 60 °C and 35 s (extension) at 72 °C for 33 cycles. PCR products were separated in 1.5% agarose gel and visualized after ethidium bromide staining.
Growth inhibition of RCC cell lines by PPARγ agonists The effect of PPARγ ligands on cell proliferation of RCC cells was determined using MTT assay[13]. Briefly, cells of 0.5×l04 cells/well were inoculated into a 96-well plate (Costar, Cambridge, MA, USA), treated with pioglitazone or troglita-zone at various concentrations. After an incubation for 24 h, 20 µL/well 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, 5 g/L) was added to each well, the medium was then removed, and 200 µL of 0.04 mol/L HCl in isopropanol was added to dissolve the reduced formazan product. The plate was read in a microplate reader (model 3550, BIO-RAD, Richmond, CA, USA) at 590 nm.
Flow cytometry assay RCC cell lines 786-O and A498 treated with pioglitazone (20 µmol/L) and troglitazone (30 µmol/L) for 24 h were harvested and washed with PBS. Cells of 1×106 were fixed in 75% ethanol at 4 °C for 24 h, washed in PBS, resuspended in 0.5–1.0 mL PBS containing 100 mg/L RNase A at 37 °C for 30 min, incubated in 10 mg/L propidium iodide in dark at 4 °C for 30 min, and sorted in a Coulter EPICS-XL Cytometer (Becton Dickinson and Beckman-Coulter, San Jose, CA, USA).
Apoptosis assay Cytosolic histon-bound DNA fragments caused by apoptosis were detected using a cell death ELISA kit (Roche Applied Science, Mannheim, Germany) according to manufacturer’s protocol. Briefly, 1×104 cells were seeded in a 96-well microplate and incubated for 4 h at 37 °C. Pioglita-zone 0–100 µmol/L or troglitazone was added and incubated for 48 h. Cells were then lysed with lysis buffer for 30 minutes at 25 °C. Supernatant 20 µL was transferred into the streptavidin coated multiplate, 4 µL anti-histon-biotin and 4 µL anti-DNA-POD were added to each well, and the plate was shaken for 2 h. After being washed with incubation buffer, 100 µL ABTS (2,2'-Azino-di[3-ethylbenzthiazoline-sulfonate]) solution was pipetted into each well. The absorbance was read immediately in a microplate reader (BIO-RAD, model 3550) at 405 nm. Furthermore, apoptosis of RCC cell lines induced by 70 µmol/L troglitazone or 80 µmol/L pioglitazone was also assayed using two other methods. Morphological changes resulting from apoptosis were determined by Hoechst 33342 staining. Cells suspended in PBS were stained with 2 mg/L Hoechst33342 and observed under fluorescence microscope using a blue filter. Cells showing cytoplasmic and nuclear shrinkage and chromatin condensation or fragmentation were defined as apoptotic cells. DNA fragmentation manifested as laddering in agarose gel was also examined. After drug treatment, the cells pellet was lysed in 10 mmol/L Tris-HCl pH 8.0, 150 mol/L NaCl, 10 mmol/L edetic acid and 0.5% SDS on ice for 10 min, and treated with RNase A and proteinase K for 1 h. Following DNA precipitation with ethanol and ammonium acetate at -20 °C for 10 h, DNA was dissolved in TE buffer, electrophoresed in 1.8% agarose gel, and visualized under UV light.
Western blot analysis Cells before and after drug treatment were extracted with lysis buffer containing 50 mmol/L Tris-HCL, pH 8.0, 150 mmol/L sodium chloride, 1% TritonX-100, 0.02% sodium azide, 100 mg/L phenylmethylsulfony fluoride and 1 mg/L aprotinin. The protein content in cell lysate was determined by bicichoninic acid assay using bovine serum albumin as the standard. Cell lysate containing 50–60 µg protein was resolved by SDS-polyacrylamide gel electrophoresis and electrophoretically transferred onto a polyvin-ylidene difluoride membrane (Millipore, Bedford, MA, USA). The membrane was blotted by antibodies against human PPARγ (1:500), proliferating cell nuclear antigen (PCNA; 1:1500), pRb (1:1000), p21 (1:1000), p27 (1:1000), cyclinD1 (1:1000), Cdk4 (1:1000), Bcl-2 (1:1000) and Bax (1:800) (Santa Cruz Biotechnology). Blotted antibody was developed by horseradish peroxidase-conjugated secondary antibody and enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Uppsala, Sweden).
Statistical analysis The experimental results shown were repeated twice or three times, unless otherwise indicated. Results are expressed as mean±SD. Statistical analysis was carried out using Student’s t-test and one-way ANOVA. Significance was set at P<0.05. Statistical analyses were performed using SPSS10.0 (SPSS Inc, Chicago, IL, USA)
Results
PPARγ expression in human RCC tissue and RCC cell lines Using RT-PCR and Western blot analysis PPARγ mRNA expression was detected in 15 human RCC tissues, 10 normal kidney tissues, 2 RCC cell lines (786-O and A498) and 2 normal kidney cell lines (HK-2 and HMCL). PPARγ expression at both mRNA (Figure 1A) and protein level (Figure 1B) was detected in all tissues and cell lines examined. Much higher PPARγ expression levels were observed in human RCC tissues and cell lines (786-O and A498) compared to normal renal tissues and cell lines (Figure 1A, 1B). Immunostaining analysis further demonstrated that PPARγ immunoreactivity was much higher over cancer cells in human RCC tissues compared to adjacent normal tissues and normal renal tissues (Figure 1C).
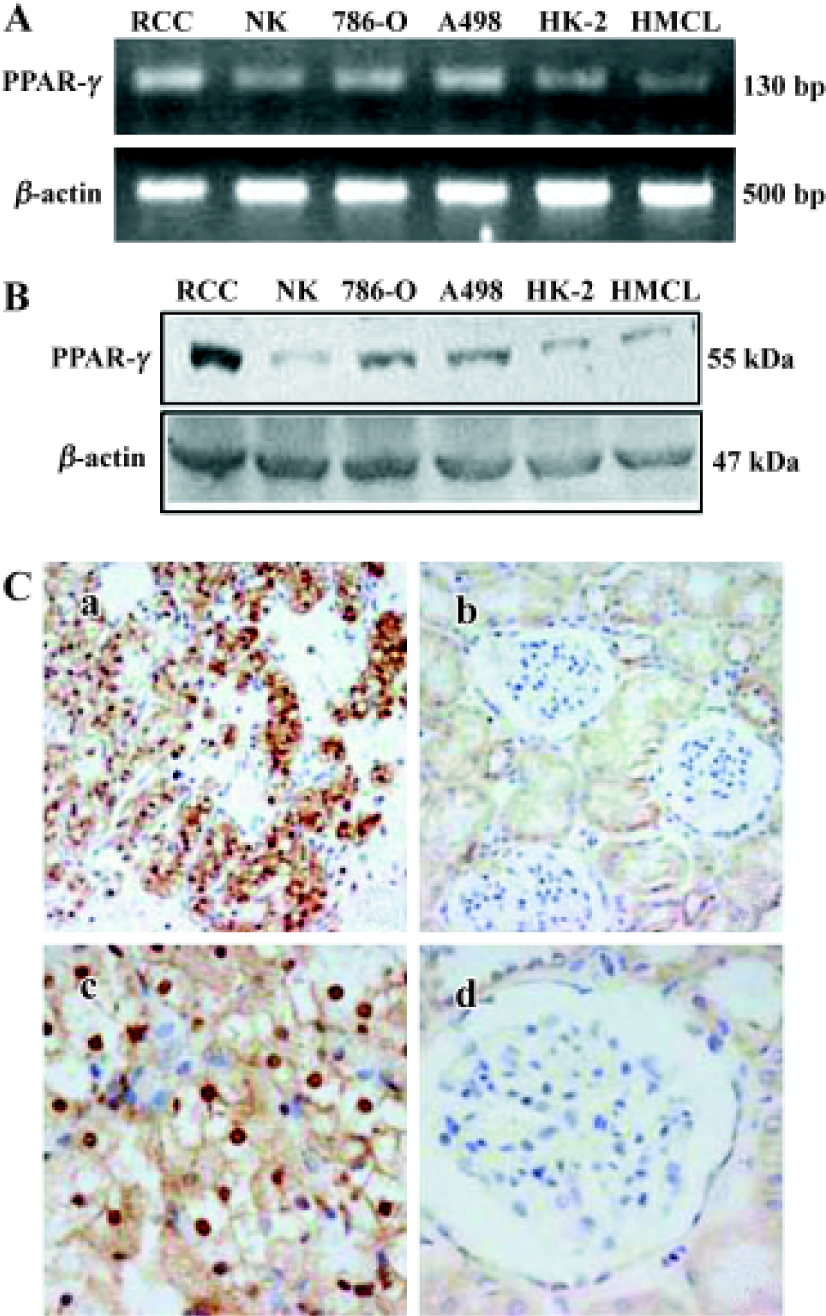
Strong staining of PPARγ was observed in 118 out of 120 human RCC tissues. PPARγ protein was predominantly localized to nucleus and cytoplasm areas of the cells. In contrast, although 16 out of the 20 normal renal samples showed PPARγ protein expression, only weak PPARγ expression was observed in cytoplasm of the medullary collecting duct cells. Statistically, the intensity of PPARγ immunoreactivity was much higher in human RCC tissues than in normal renal tissues (2.16±0.85 vs 0.90±0.73; P< 0.01).
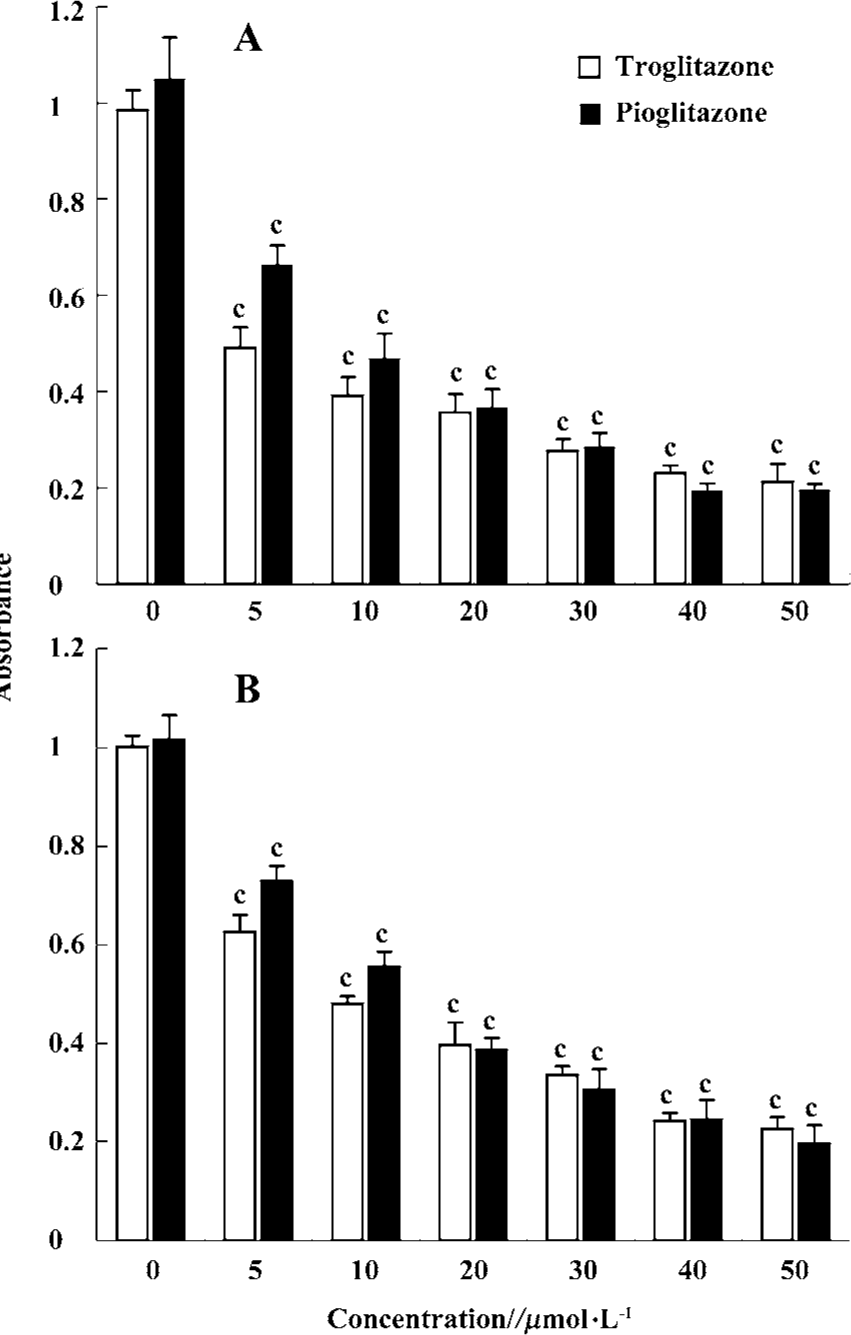
PPARγ agonists inhibited proliferation of RCC cell lines To determine the effect of PPARγ ligands on growth of human RCC cell lines, the exponentially growing 786-O and A498 cells were treated with 0–50 µmol/L pioglitazone and troglitazone for 24 h. Cell growth was determined by an MTT assay. Figure 2 showed that two TZD PPARγ agonists pioglitazone and troglitazone significantly inhibited the cell growth of RCC in a dose-dependent manner, with the calculated IC50 values of 7.08 and 11.32 µmol/L for pioglitazone and 5.71 and 8.38µmol/L for troglitazone in 786-O and A498 cells, respectively.
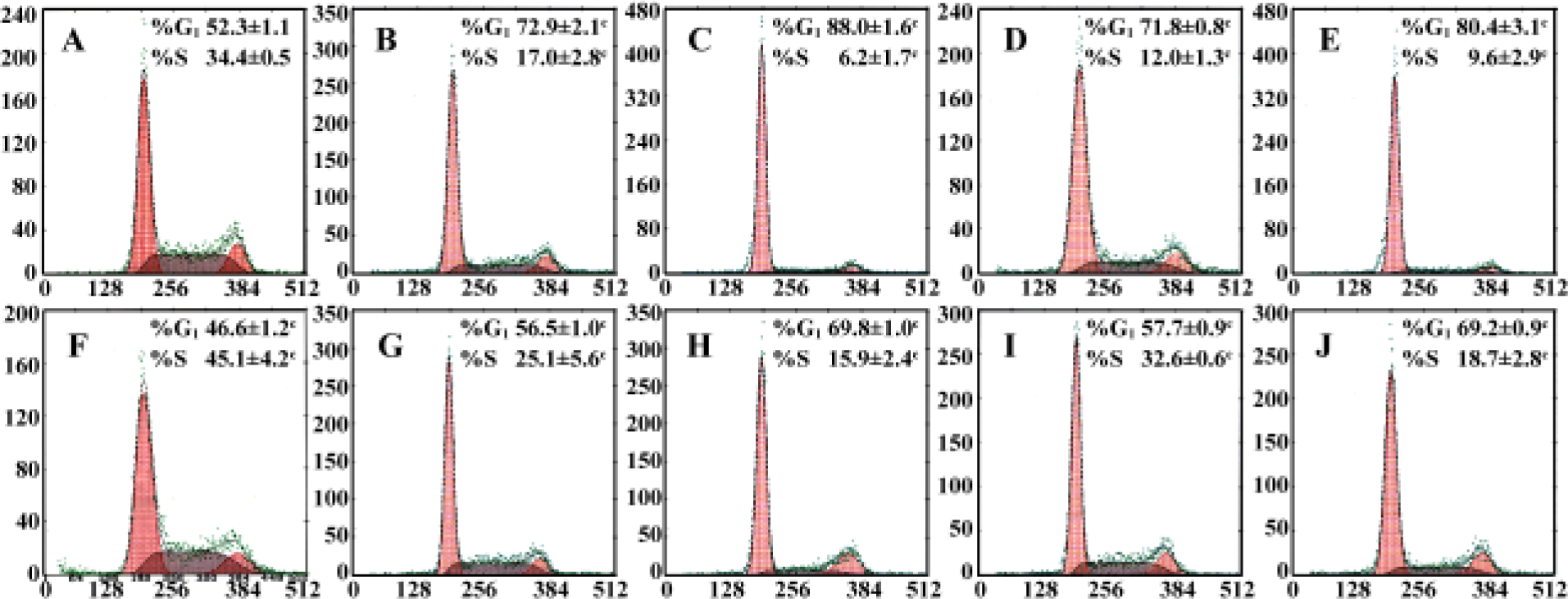
Effect of PPARγ agonists on cell cycle of RCC cell lines Cell cycle analysis was performed in two RCC cell lines after exposure to 20–30 µmol/L pioglitazone and troglitazone for 24 h. In both cell lines, pioglitazone and troglitazone caused a dose-dependent increase in the G0/G1 phase and decrease in the S phase (Figure 3).
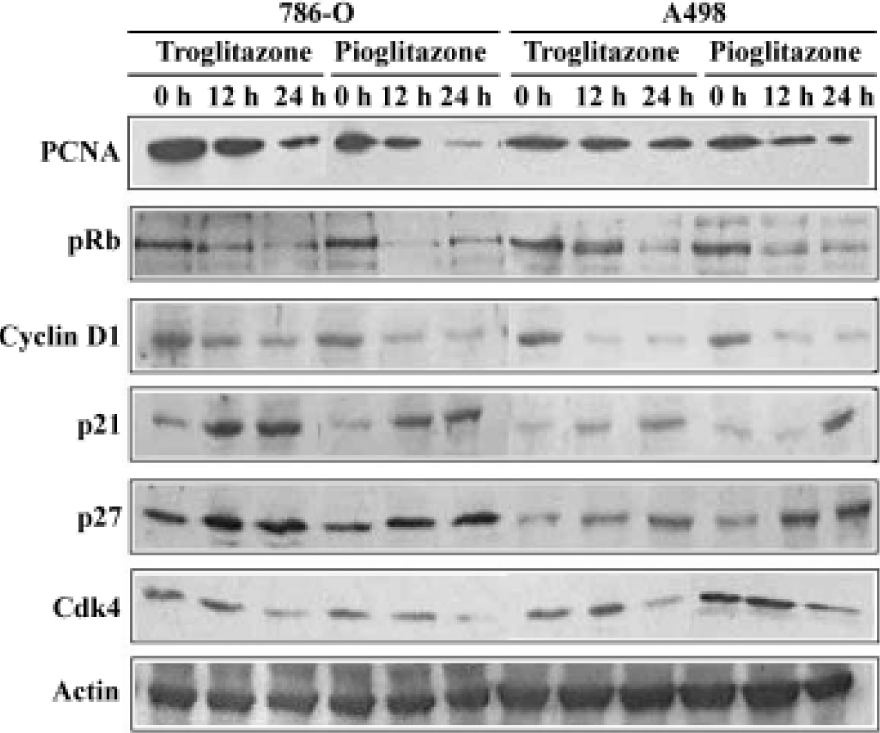
Effect of PPARγ ligands on cell cylce protein expression in human RCC cell lines The cell cycle is tightly regulated through a complex network of positive and negative regulatory molecules including cyclin dependent kinases (Cdks), cyclins, and Cdk inhibitors (Cdkis). To elucidate the role of these molecules in the inhibition of cell cycle induced by pioglitazone and troglitazone in RCC cell lines, protein extract was prepared from the cells treated with 30 µmol/L pioglitazone or troglitazone for 0, 12, and 24 h. Immuno-blotting was performed using antibodies against human PCNA, pRb, cyclin D1, p21, p27, and Cdk4. As shown in Figure 4, pioglitazone and troglitazone treatment time-dependently caused a marked decrease in PCNA, pRb, Cyclin D1 and Cdk4 expression and a significant increase in p21 and p27 expression.
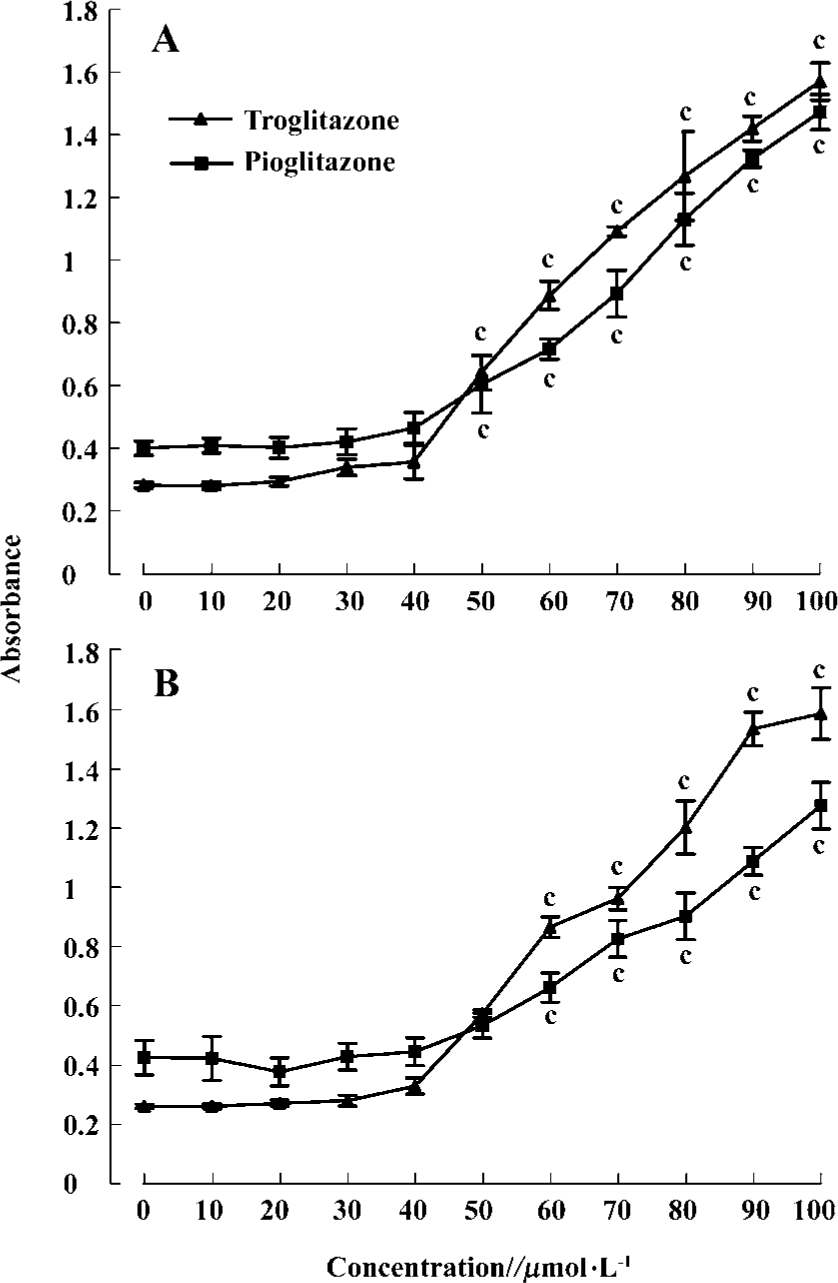
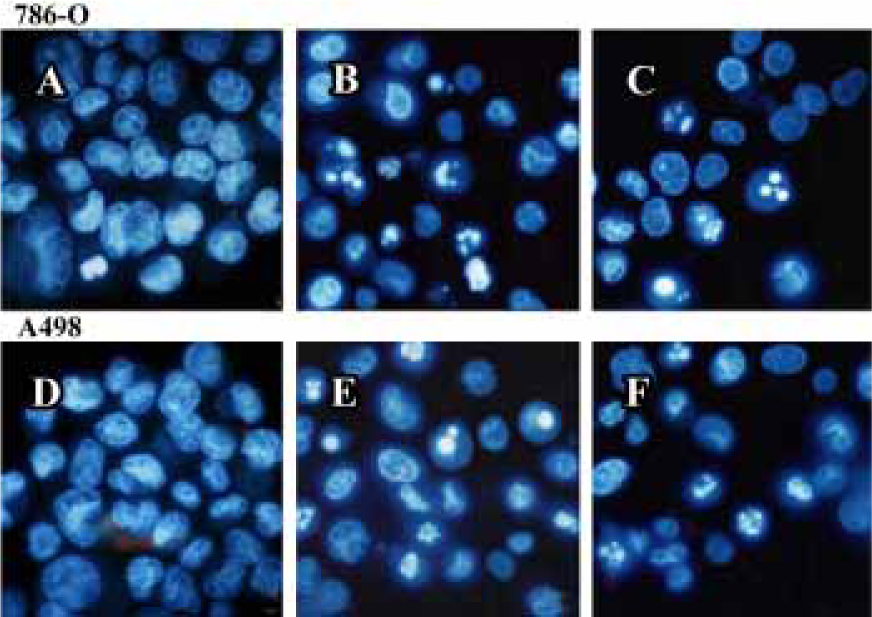
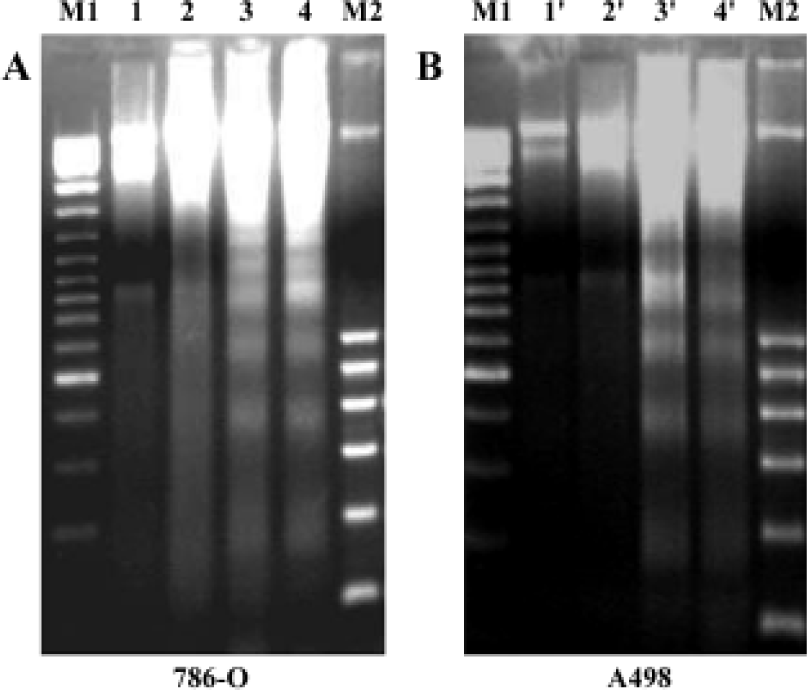
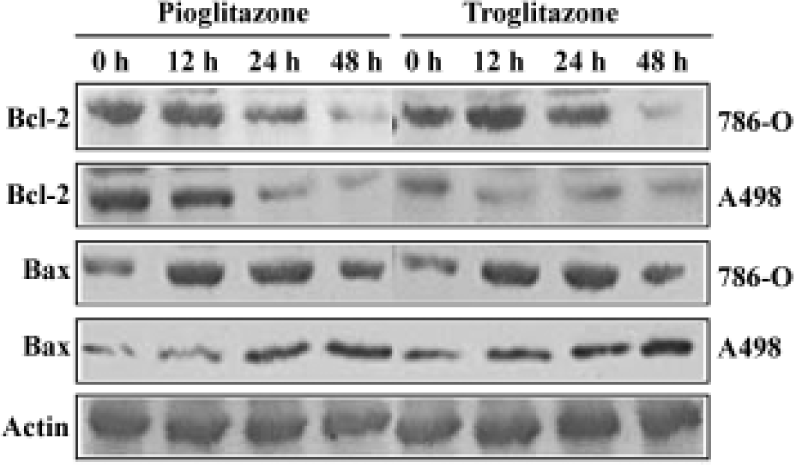
PPARγ agonist induced apoptosis in human RCC cell lines The ability of pioglitazone and troglitazone to induce apoptosis in two RCC cell lines was initially determined by the cytosolic histon-bound DNA fragments and cell death ELISA assay. We found that pioglitazone and troglitazone treatment for 48 h significantly increased apoptotic cell numbers in a dose-dependent manner, with IC50 values of 67.73 µmol/L and 78.12 µmol/L for pioglitazone and 65.11 µmol/L and 63.91µmol/L for troglitazone in 786-O and A498 cells, respectively (Figure 5). When 786-O and A498 cells were exposed to either 80 µmol/L pioglitazone or 70 µmol/L troglitazone for 48 h, they exhibited typical morphological changes of apoptosis including cytoplasmic and nuclear shrinkage, chromatin condensation and fragmentation after staining with Hoechst 33342 (Figure 6). The genomic DNA of treated cells exhibited a characteristic ladder pattern after electrophoresis in agarose gel (Figure 7). To further explore the mechanisms involved in PPARγ agonist-induced apoptosis, the expression of proteins involved in the Bcl-2 apoptotic pathway was examined in RCC cell lines. Treatment of RCC cells with pioglitazone or troglitazone for 48 h decreased the levels of Bcl-2 expression in a time-dependent manner in both 786-O and A498 cells. In contrast, the expression of Bax was increased following TZD treatment(Figure 8).
Discussion
PPARγ is a nuclear receptor transcription factor and plays an important role in many biological processes including adipogenesis, cell growth regulation, and cell differentiation. The synthetic TZD class of compounds, including pioglita-zone and troglitazone, are originally developed as therapeutic agents for diabetes mellitus and have been recently found to be specific ligands for PPARγ with high affinity. In the past decade, evidence began to emerge suggesting TZD PPARγ agonists not only exert antidiabetic effect in type II diabetes mellitus but also induce cell growth arrest, apoptosis and terminal differentiation in many human malignant tumors including colon cancer, breast cancer, pituitary adenomas, pancreatic carcinoma and esophageal cancer[9–11,19,20], suggesting PPARγ may be a potential therapeutic target for treatment of certain human cancers. The present study provides evidence that high expression of PPARγ was observed in human RCC tissues and cell lines, and activation of PPARγ resulted in G0/G1 cell cycle arrest and apoptosis involved Bcl-2 pathway.
In the present study, we observed that PPARγ was abundantly expressed in almost all high-grade human RCC specimens (98.3%), and two RCC cell lines were examined. Its intracellular distribution was mainly localized in nuclear area. In contrast, although PPARγ immunostaining was also positive in most normal renal tissues, its expression was much lower than that in RCC tissues and limited in medullary collecting duct cells in the kidney. Furthermore, its subcellular localization was predominantly in the cytoplasm. The differences of PPARγ in quantity and intracellular distribution between human RCC and normal kidney tissues may implicate an important role of PPARγ in tumorigenesis of renal cell carcinoma.
Consistent with a previous study[21], activation of PPARγ in two human RCC cell lines resulted in inhibition of RCC cell growth as assessed using MTT assay and Flow Cytometry. As seen in other tumor cells, treatment of RCC cells lines with pioglitazone and troglitazone for 24 h caused G0/G1 phase arrest and blocked cells from entering the S phase[13,19,22–24]. To explore the mechanism involved in PPARγ-induced cell growth arrest, RCC cells were treated with pioglitazone and troglitazone and expression of cell cycle proteins was examin-ed. We observed a marked reduction in PCNA, pRb, Cdk4 and Cyclin D1 expression and a dramatic increase in p21 and p27 expression in TZD-treated RCC cells. Down-regulation of PCNA, a nuclear protein essential for DNA replication and repair by DNA polymerase δ[25], is in agreement to the low proliferation rate observed in these treated RCC cells. Proliferation of eukaryotic cells is tightly regulated by expression and sequential activation of cell cycle-dependent cyclins, Cdks and CdkIs[26]. pRb, a key regulator of G1 cell cycle progression, is phosphorylated by a set of Cyclin-Cdk complexes, such as the complexes of Cyclin D and Cdk4/6 or cyclin E and Cdk2[27,28]. In its dephosphorylated state, pRb binds to E2F and inhibits G1-S phase transi-tion. In contrast, phosphorylation of pRb causes breakdown of E2F/pRb complex, and initiates DNA synthesis. In the present study, Decreased expression of phosphorylated pRb was found in these treated cells. CdkIs are cell cycle regulatory molecules having negative effects on cell cycle machinery by binding to various cyclin-Cdk complexes and inhibiting their activities. In mammals, there are two structurally defined classes of CdkIs, the INK4 family and the KIP/CIP family[29]. The KIP/CIP family, including p21CIP, p27KIP1, and p57KIP2, interacts with cyclin E-Cdk2, cyclin D-Cdk4, cyclin D-Cdk6, and cyclin A-Cdk2 complexes, and inhibits their activities[28]. The decrease of cyclin D1 and Cdk4 and increased expression of CdkI proteins including p21 and p27 strongly suggest TZD PPARγ agonists induced the overexpression of the members of KIP/CIP family which sequentially inhibited the activities of cyclin D1-Cdk4 complex, the phosphorylation of pRb, and the G1-S phase transition. These findings are consistent with the observation that two TZD PPARγ agonists decreas-ed cell proliferation in MTT assay and arrested cells in G0/G1 phase by flow cytometry.
Cell cycle status and cell programmed death are usually closely associated[13]. Besides the cell cycle arrest, the inhibition of cell growth observed in RCC cells treated with pioglitazone and troglitazone may also be a result of the increase in apoptosis[30]. Cells failing to progress to mitosis phase are destined for apoptosis. TZD have been reported to be potent agents in inducing apoptosis in many human tumors including breast cancer, pituitary tumors and gastric cancer[10–12]. In the present study, we clearly demonstrated that at high concentrations PPARγ agonists pioglitazone and troglitazone caused marked apoptosis in human RCC cells as assessed by morphological change, DNA fragmentation and the cell death ELISA assay. These studies suggest that TZD may exert pro-apoptotic effect on human RCC cells.
Multiple pathways are involved in apoptosis including Bcl-2 system, fas/fasL pathway, and caspase cascade[31–33]. To explore the molecular mechanism through which TZD PPARγ agonist induces apoptosis in human RCC cells, we examined Bcl-2 and Bax expression following pioglitazone and troglitazone treatment in 786-O cells and A498 cells. We observed for the first time that activation of PPARγ by both TZD markedly decreased Bcl-2 protein expression but increased Bax protein expression. Because Bcl-2 protects cells from apoptosis, while the increase of Bax induces apopto-sis[34,35], our findings suggest that decreased Bcl-2 expression and increased Bax expression may participate in TZD PPARγ agonist-induced apoptosis in human RCC cells.
PPARγ agonists, such as 15-dPGJ2, troglitazone, have been shown to possess effects not involving PPARγ[36,37], For instance, the potencies for inhibition of cholesterol synthesis by troglitazone is mechanistically distinct from the transcriptional regulation by PPARγ[38] . In the present study, low doses of TZD fail to induce RCC cell death. It has been reported that TZD promote T cell survival at doses that induce optimal PPARγ transcriptional activity[39], and cell deaths induced by TZD always use concentrations of TZD several orders of magnitude higher than KD for PPARγ[39,40]. More-over, troglitazone could induce apoptosis of rat hepatoma cells, but rosiglitazone, a potent PPARγ agonist, did not have the same effect[41], indicating that PPARγ independent pathways, such as TZD-induced loss of mitochondrial membrane potential[42], may be involved in the mechanisms of TZD induced cell apoptosis. Furthermore, it has also been reported that PPARγ agonists could induce apoptosis in non-malignant cells, including isolated rat mesangial cells[43]. Thus the mechanisms of anticaner effect of TZD need to be further explored.
In summary, the present study provides evidence that PPARγ is highly expressed in human renal cell carcinoma cells. Activation of PPARγ by TZD PPARγ agonists results in inhibition of cell proliferation, likely by arresting the cells in G0/G1 cell cycle phase. In addition, PPARγ agonists also induce marked apoptosis partly through decreased Bcl-2 and enhanced Bax protein expression in human RCC cells. Additional research will need to be performed to explore the mechanism of TZD which induced cell apoptosis, and the potential therapeutic value of PPARγ agonists in renal cell carcinoma.
References
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990;347:645-50.
- Schoonjans K, Martin G, Staels B, Auwerx J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr Opin Lipidol 1997;8:159-66.
- Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signaling pathways through heterodimer formation of their receptors. Nature 1992;358:771-4.
- Kliewer SA, Lehmann JM, Milburn MV, Willson TM. The PPARs and PXRs: nuclear xenobiotic receptors that define novel hormone signaling pathways. Recent Prog Horm Res 1999;54:345-67.
- Palmer CN, Hsu MH, Griffin HJ, Johnson EF. Novel sequence determinants in peroxisome proliferator signaling. J Biol Chem 1995;270:16114-21.
- Camp HS, Ren D, Leff T. Adipogenesis and fat-cell function in obesity and diabetes. Trends Mol Med 2002;8:442-7.
- Rangwala SM, Lazar MA. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol Sci 2004;25:331-6.
- Fauconnet S, Lascombe I, Chabannes E, Adessi GL, Desvergne B, Wahli W, et al. Differential regulation of vascular endothelial growth factor expression by peroxisome proliferator-activated receptors in bladder cancer cells. J Biol Chem 2002;277:23534-43.
- Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med 1998;4:1046-52.
- Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptor-gamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci USA 1998;95:8806-11.
- Heaney AP, Fernando M, Melmed S. PPAR-gamma receptor ligands: novel therapy for pituitary adenomas. J Clin Invest 2003;111:1381-8.
- Takahashi N, Okumura T, Motomura W, Fujimoto Y, Kawabata I, Kohgo Y. Activation of PPARgamma inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett 1999;455:135-9.
- Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD. Expression of peroxisome proliferator-activated receptor gamma (PPARgamma) in human transitional bladder cancer and its role in inducing cell death. Neoplasia 1999;1:330-9.
- Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, et al. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-γ ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci USA 1999;96:3951-6.
- Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci USA 2000;97:10990-5.
- Golimbu M, Joshi P, Sperber A, Tessler A, Al-Askari S, Morales P. Renal cell carcinoma: survival and prognostic factors. Urology 1986;27:291-301.
- Flanigan RC, Campbell SC, Clark JI, Picken MM. Metastatic renal cell carcinoma. Curr Treat Options Oncol 2003;4:385-90.
- Guan Y, Zhang Y, Schneider A, Davis L, Breyer RM, Breyer MD. Peroxisome proliferator-activated receptor-gamma activity is associated with renal microvasculature. Am J Physiol Renal Physiol 2001;281:1036-46.
- Motomura W, Okumura T, Takahashi N, Obara T, Kohgo Y. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human pancreatic carcinoma cells. Cancer Res 2000;60:5558-64.
- Hashimoto Y, Shimada Y, Itami A, Ito T, Kawamura J, Kawabe A, et al. Growth inhibition through activation of peroxisome proliferator-activated receptor gamma in human oesophageal squamous cell carcinoma. Eur J Cancer 2003;39:2239-46.
- Inoue K, Kawahito Y, Tsubouchi Y, Kohno M, Yoshimura R, Yoshikawa T, et al. Expression of peroxisome proliferator-activated receptor gamma in renal cell carcinoma and growth inhibition by its agonists. Biochem Biophys Res Commun 2001;287:727-32.
- Heaney AP, Fernando M, Yong WH, Melmed S. Functional PPAR-gamma receptor is a novel therapeutic target for ACTH-secreting pituitary adenomas. Nat Med 2002;8:1281-7.
- Mossner R, Schulz U, Kruger U, Middel P, Schinner S, Fuzesi L, et al. Agonists of peroxisome proliferator-activated receptor gamma inhibit cell growth in malignant melanoma. J Invest Dermatol 2002;119:576-82.
- Brockman JA, Gupta RA, Dubois RN. Activation of PPARgamma leads to inhibition of anchorage-independent growth of human colorectal cancer cells. Gastroenterology 1998;115:1049-55.
- Luo Y, Hurwitz J, Massague J. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature 1995;375:159-61.
- Pines J. Four-dimensional control of the cell cycle. Nat Cell Biol 1999;1:73-9.
- Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway in cell cycle control and cancer. Exp Cell Res 1997;237:1-6.
- Sherr CJ. Mammalian G1 cyclins. Cell 1993;73:1059-65.
- Aprelikova O, Xiong Y, Liu ET. Both p16 and p21 families of cyclin-dependent kinase (CDK) inhibitors block the phosphorylation of cyclin-dependent kinases by the CDK-activating kinase. J Biol Chem 1995;270:18195-7.
- Shi C, Yu L, Yang F, Yan J, Zeng H. A novel organoselenium compound induces cell cycle arrest and apoptosis in prostate cancer cell lines. Biochem Biophys Res Commun 2003;309:578-83.
- Zhang CL, Wu LJ, Tashiro S, Onodera S, Ikejima T. Oridonin induces apoptosis of HeLa cells via altering expression of Bcl-2/Bax and activating caspase-3/ICAD pathway. Acta Pharmacol Sin 2004;25:691-8.
- Filippova M, Parkhurst L, Duerksen-Hughes PJ. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J Biol Chem 2004;279:25729-44.
- Orrenius S. Mitochondrial regulation of apoptotic cell death. Toxicol Lett 2004;149:19-23.
- Li XH, Li JJ, Zhang HW, Sun P, Zhang YL, Cai SH, et al. Nimesulide inhibits tumor growth in mice implanted hepatoma: overexpression of Bax over Bcl-2. Acta Pharmacol Sin 2003;24:1045-50.
- Salomons GS, Brady HJ, Verwijs-Janssen M, Van Den Berg JD, Hart AA, Van Den Berg H, et al. The Bax alpha:Bcl-2 ratio modulates the response to dexamethasone in leukaemic cells and is highly variable in childhood acute leukaemia. Int J Cancer 1997;71:959-65.
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 2001;7:48-52.
- Davies GF, Khandelwal RL, Wu L, Juurlink BH, Roesler WJ. Inhibition of phosphoenolpyruvate carboxykinase (PEPCK) gene expression by troglitazone: a peroxisome proliferator-activated receptor-gamma (PPARgamma)-independent, antioxidant-related mechanism. Biochem Pharmacol 2001;62:1071-9.
- Wang M, Wise SC, Leff T, Su TZ. Troglitazone, an antidiabetic agent, inhibits cholesterol biosynthesis through a mechanism independent of peroxisome proliferator-activated receptor-gamma. Diabetes 1999;48:254-60.
- Wang YL, Frauwirth KA, Rangwala SM, Lazar MA, Thompson CB. Thiazolidinedione activation of peroxisome proliferator-activated receptor gamma can enhance mitochondrial potential and promote cell survival. J Biol Chem 2002;277:31781-8.
- Yu C, Chen L, Luo H, Chen J, Cheng F, Gui C, et al. Binding analyses between human PPARgamma-LBD and ligands. Eur J Biochem 2004;271:386-97.
- Bae MA, Rhee H, Song BJ. Troglitazone but not rosiglitazone induces G1 cell cycle arrest and apoptosis in human and rat hepatoma cell lines. Toxicol Lett 2003;139:67-75.
- Bova MP, Tam D, McMahon G, Mattson MN. Troglitazone induces a rapid drop of mitochondrial membrane potential in liver HepG2 cells. Toxicol Lett 2005;155:41-50.
- Tsuchiya T, Shimizu H, Shimomura K, Mori M. Troglitazone inhibits isolated cell proliferation, and induces apoptosis in isolated rat mesangial cells. Am J Nephrol 2003;23:222-8.