
Transforming growth factor-β1 gene polymorphisms associated with chronic obstructive pulmonary disease in Chinese population1
Introduction
Chronic obstructive pulmonary disease (COPD) is currently the leading cause of disability-adjusted life years. Globally, it is projected to be the 3rd most important cause of death by the year 2020, and it is estimated that the national prevalence is up to 3.1% in the Chinese adult population. Cigarette smoking is the major risk factor for COPD; however, only approximately 15% of smokers develop clinically relevant airflow obstruction[1]. This variation in the susceptibility to cigarette smoke in combination with the familiar aggregation of COPD suggests that there may be a genetic component to the development of COPD. Multiple studies in diverse populations have shown evidence for a large genetic contribution to the variability in pulmonary function and for the familial aggregation of COPD patients[2,3]. As expected, segregation analysis suggests that multiple genes may be involved. At present, however, only a single gene, α1-antitrypsin, a potent inhibitor of inflammatory cell protease in the lung, has been unequivocally implicated in the development of COPD. The association between the ZZ type polymorphism of this gene and COPD has been established[4]. Furthermore, the associations between COPD and polymorphisms in several other genes of potential importance to COPD pathogenesis also have been studied. These include α1-antichymotrypsin[5], microsomal epoxide hydrolase[6], vitamin D-binding protein[7], and tumor necrosis factor-α[8].
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that regulates the proliferation and differentiation of a wide variety of cell types in vitro. In humans, the TGF-β family includes 3 isoforms (TGF-β1, TGF-β2, and TGF-β3) with great structural and functional similarities[9]. TGF-β1 is the most abundant isoform and is highly conserved in primary sequence through evolution. It is synthesized as precursor latent forms (390 amino acids long), which have no known biological activity[10]. The active form of TGF-β1 is released proteolytically from the precursor form[11] and consists of a disulphide-linked 25 kDa homodimer of two 112 amino acid peptides. The coding region of the TGF-β1 gene consists of 7 exons and 6 introns[12].
The expression of TGF-β1 is influenced by polymorphisms in the TGF-β1 gene, and some of these polymorphisms may be associated with COPD and other diseases[13–15]. Wu et al investigated the association between the single nucleotide polymorphism T+869C (Leu10Pro) at exon 1 of the TGF-β1 gene with COPD in a Caucasian Americans white popula-tion[16]. There are several registered polymorphisms within the TGF-β1 gene that might be functional and might be associated with COPD. In particular, a promoter polymorphism at -509 C/T is associated with the diagnosis of asthma and may enhance TGF-β1 gene transcription[17], but whether there is an association between the -509 C/T polymorphism and COPD has not yet been investigated. The aim of present study was to screen for DNA sequence variants in the TGF-β1 promoter, to establish the prevalence of each genotype in individuals with COPD and controls in a Chinese population, and to identify any haplotypes associated with a predisposition to COPD.
Materials and methods
Subjects Eighty-four patients with COPD were recruited from the West China Hospital, Sichuan University, Chengdu, China. The definition of COPD was consistent with that in the American Thoracic Society (ATS) consensus statement. The patients had a history of chronic or recurrent productive cough for >2 years and decreased maximum expiratory flow, which had been slowly progressive and irreversible. The presence of other lung or cardiac diseases as the cause of patient symptoms was excluded by clinical and radiographic examinations. The criteria for enrolment were as follows: (i) individuals with a forced expiratory volume in 1 s (FEV1)<70% of predicted, an FEV1/forced vital capacity (FVC) ratio of <70%, and an increase in FEV1 of <12% 15 min after the inhalation of 400 µg Fenoterol HBr MDI (Berotec; Boehringer Ingelheim, Ridgefield, CT, USA); and (ii) patient consent to participate in the study. Most of the patients were receiving oral methylxanthine, inhaled anticholinergic agents, and inhaled β2-agonists as needed. The patient’s name, age, sex, family history, smoking habits, the number of cigarettes smoked, the duration of diseases, and chest radiographic findings were recorded. Pulmonary function testing (CHESTAC-33-8800, Chest Ltd, Tokyo, Japan) was performed according to the ATS performance requirements. Ninety-seven unrelated, age-matched healthy subjects, who had no known medical illness or family disorders and were not taking any medications, acted as control subjects. This study was approved by the Internal Review Board of the West China Hospital, Sichuan University, and signed informed consent forms were obtained from all subjects.
DNA preparation and polymerase chain reaction amplification Genomic DNA was prepared from peripheral blood leukocytes using the “salting-out”[18] procedure and stored at 4 °C. Fragments containing the single nucleotide polymorphism (SNP) G-800A or T-509C were amplified by polymerase chain reaction (PCR). The primer sequence was based on the DNA sequence from GenBank accession numbers NM_000660 and X05839. For the detection of polymorphism at nucleotide acid -800, the primer pairs were 5'-CTGGCAGT-TGGCGAGAACA-3' (sense) and 5'-TAGAAAGGACAGAA-GCGGTG-3' (antisense); and the generated PCR product size was 326 base pairs (bp). The primer pairs for delineating the polymorphism at nucleotide acid -509 were 5'-CCAGCTAA-GGCATGGC ACCG-3' (sense) and 5'-GCGGTGTGGGTCA-CCAGAGA-3' (antisense); the PCR product size from these primers was 300 bp. Each PCR amplification mixture contained 0.1 µg genomic DNA, 0.2 µmol/L primers, 50 µmol/L deoxyribonucleotide triphosphate (dNTP), and standard PCR buffer in a total volume of 50 µL. The reaction mixture was preheated at 94 °C for 4 min. Subsequently, 0.4 units of Taq polymerase was added. The 30 cycles of PCR amplification were performed with a temperature profile consisting of denaturation at 94 °C for 45 s, annealing at 58 °C and 61 °C for 30 s, extension at 72 °C for 30 s, and final extension at 72 °C for 10 min. The reactions were carried out in a PTC-200 Programmable Thermal Controller (MJ Research, Watertown, MA, USA).
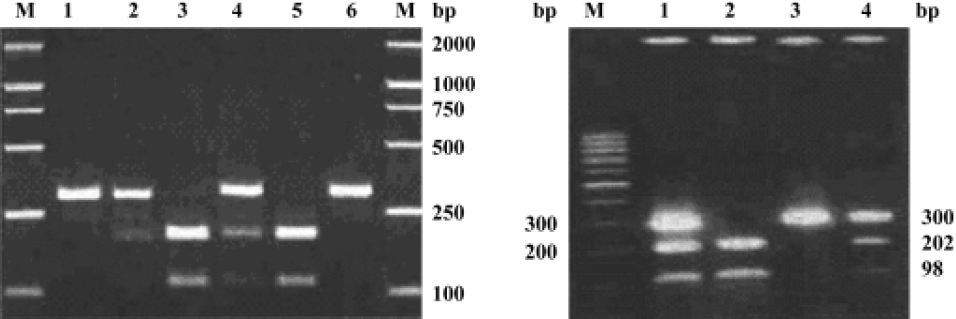
Genotyping Genotyping for the -800G/A and -509C/T polymorphisms was performed by digestion of PCR product with the restriction enzymes MaeIII and SauI (Amersham Biosciences, Piscataway, NJ, USA), respectively. After PCR, 10 µL of the reaction mixture was digested with 1 U MaeIII or SauI in 1×buffer M (Amersham Biosciences) for 3 h at 37 ºC. The digest mixture was resolved on a 2.0% agarose gel stained with ethidium bromide. For the -800G/A polymo-rphism, DNA from individuals with the homozygous G genotype (GG) produced 1 band at 326 bp; the homozygous A genotype (AA) produced 2 bands, 1at 206 bp and 1 at 120 bp; and the heterozygous genotype (GA) produced all 3 bands. A gel of the restriction fragment length polymorphism digestion products is shown in Figure 1A. For the -509C/T polymorphism, individuals with the homozygous C genotype (CC) produced 1 band at 300 bp; the homozygous T genotype (TT) produced 2 bands, 1 at 202 bp and 1 at 98 bp; and the heterozygous genotype (CT) produced all 3 bands, as shown in Figure 1B. Control DNA samples representative of each of the different genotypes were included in each digestion. Repeat genotyping was performed on 5 of every 100 samples chosen by random selection. Genotyping errors are estimated to have occurred at a frequency of <1%.
Statistical analysis Data analyses were performed with the Statistical Package for the Social Science (SPSS, version 10.0, Inc in Chicago, Illinois, USA). Data were expressed as mean±SEM. The significance level for statistical tests was taken to be 0.05. Deviation of the genotype counts from the Hardy-Weinberg equilibrium was tested using a χ2 test with 1 degree of freedom (df). Differences between the patients with COPD and the controls with respect to the allele frequencies and genotype distributions were analyzed by χ2 test or Fisher’s exact test when necessary. Haplotype frequencies for pairs of alleles, as well as χ2 values for allele associations, were estimated by the Estimating Haplotype-frequencies software program[19–20]. Linkage disequilibrium coefficients D’=D/Dmax (D is the difference between the observed and the expected gamete frequency, Dmax is the maximum disequilibrium, which occurs when all double heterozygotes are either in linkage phase (AB/ab) or in repulsions phase (Ab/aB). were calculated by the 2LD program[21].
Results
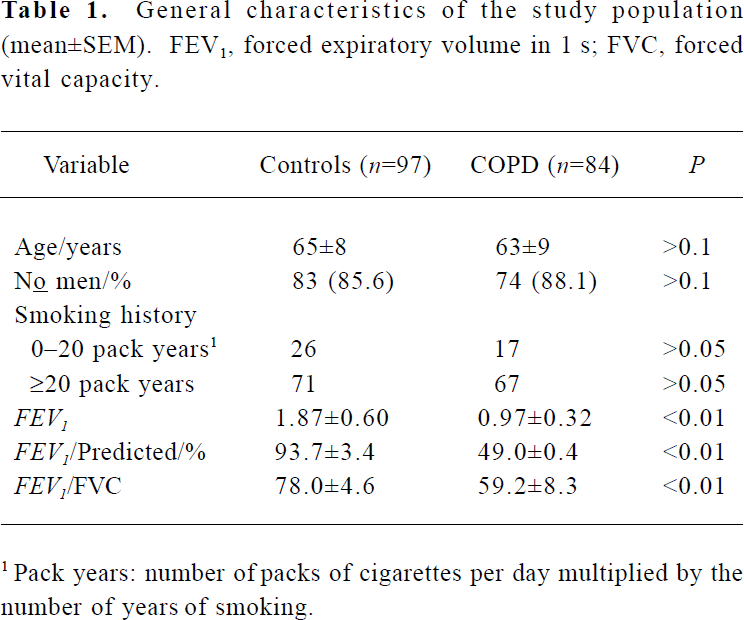
General characteristics The study population consisted of 84 patients with COPD and 97 control subjects (Table 1). The COPD cases and control subjects did not differ significantly in sex, age, and smoking history characteristics. The parameters used for FEV1, FEV1/predicted, and FEV1/FVC were significantly decreased in the COPD case subjects compared with the controls (Table 1, P<0.01).
Full table
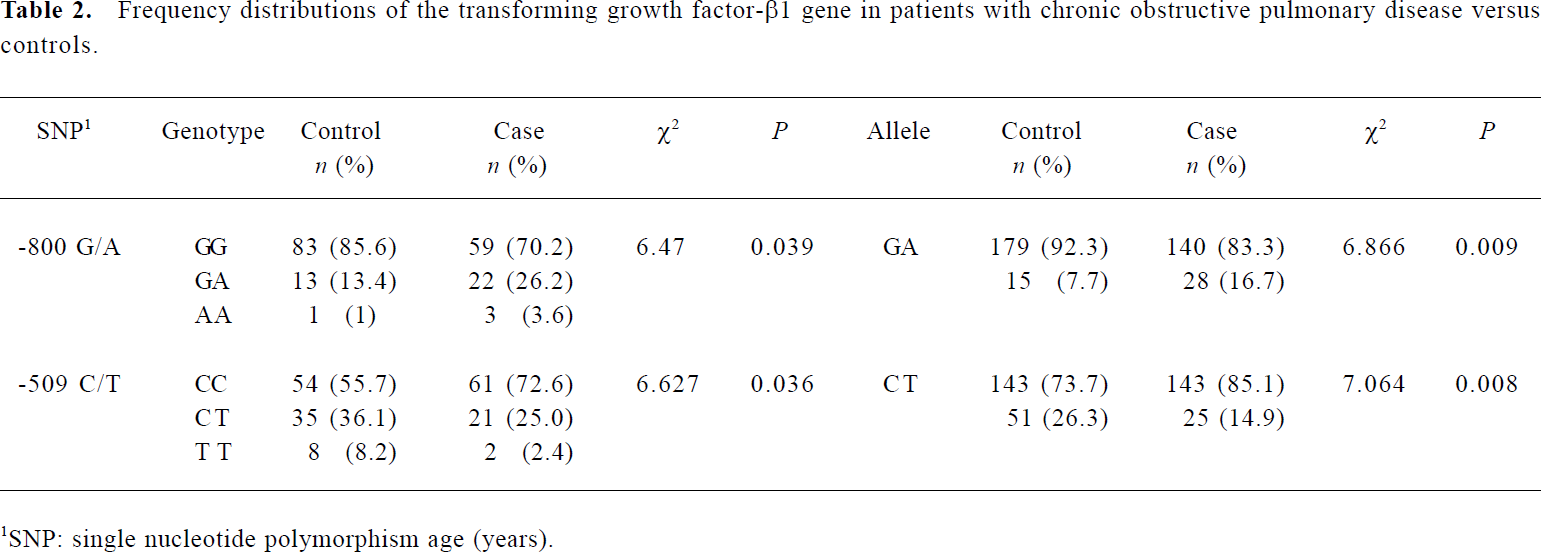
Distribution of the G-800A and C-509T in COPD patients and controls To determine the prevalence of the SNP G-800A and C-509T, we screened them in all the 84 COPD patients and in the 97 controls. The genotype distribution and allele frequencies are listed in Table 2. No deviation from Hardy-Weinberg equilibrium (for SNP G-800A, χ2=1.379, df=1, P=0.240 in the COPD group, χ2=1.737, df=1, P=0.188 in controls; for SNP C-509T, χ2=1.488, df=1, P=0.222 in the COPD group, χ2=2.400, df=1, P=0.121 in controls) was noted in the COPD or control groups. As a result, more carriers of the -800A allele, or fewer carriers of -509T allele, were detected in the COPD patients compared with in the control subjects (for the -800A allele, 29.8% vs 14.4%, χ2=6.257, df=1, P=0.012; for the -509T allele, 27.3% vs 44.3%, χ2=5.582, df=1, P=0.018). The prevalence of the -800A allele was significantly higher in COPD patients than in control subjects (P=0.009), whereas the frequency of -509T allele was significantly decreased in COPD patients compared with that in control subjects (P=0.008).
Full table
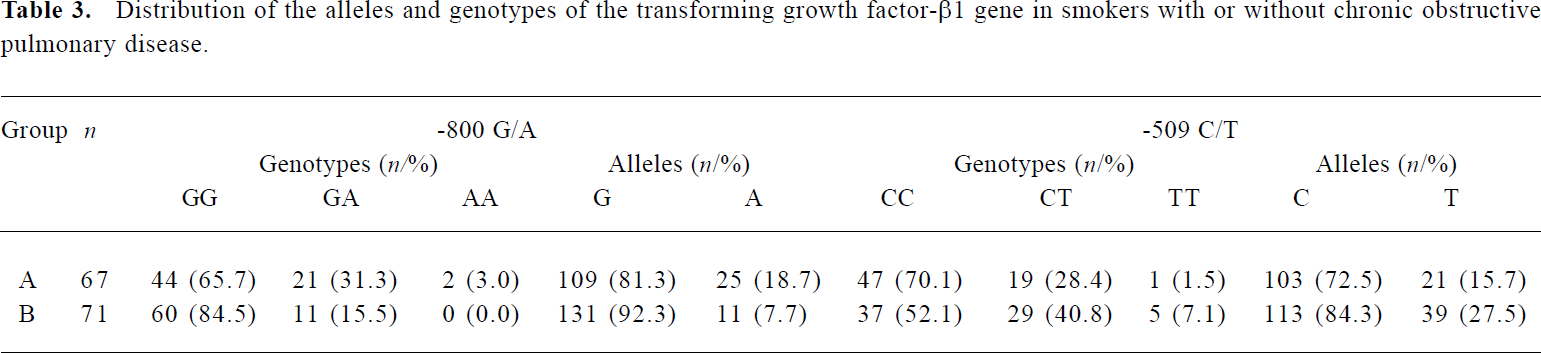
Relationship between cigarette smoking and the distribution of TGF-β1 gene polymorphisms The allele and genotype distributions were compared in heavy smokers (smoking history ≥20 pack years (number of packs of cigarettes per day multiplied by the number of years of smoking) to determine whether the prevalence of different alleles or genotypes was associated with smoking. Results showed that the frequency of the -800A allele was still significantly higher and the frequency of the -509T allele was significantly decreased in patients with COPD (group A) than in control subjects (group B) (χ2=7.235, P=0.007, and χ2=5.636, P=0.018, respectively; Table 3).
Full table
Linkage disequilibrium between -800G/A and -509C-T polymorphisms in the TGF-β1 gene We analyzed the relationship between -800G/A and -509C-T polymorphisms and their effects on COPD. The extent of D in pairwise combinations of alleles in loci at the TGF-β1 promoter was estimated by means of maximum likelihood from the frequency of diploid genotypes in the patients with asthma and controls. Haplotype frequencies and the coefficient of linkage disequilibrium (D’) are given in Table 4. It was clear that the D’ values for polymorphisms at nucleotides -800 bp and -509 bp differ significantly from zero, and the frequency of the AC haplotype, consisting of the least common base at -800 and the most common base at -509, was significantly higher in patients with COPD than in controls (0.056 vs 0.021, respectively, P<0.05).
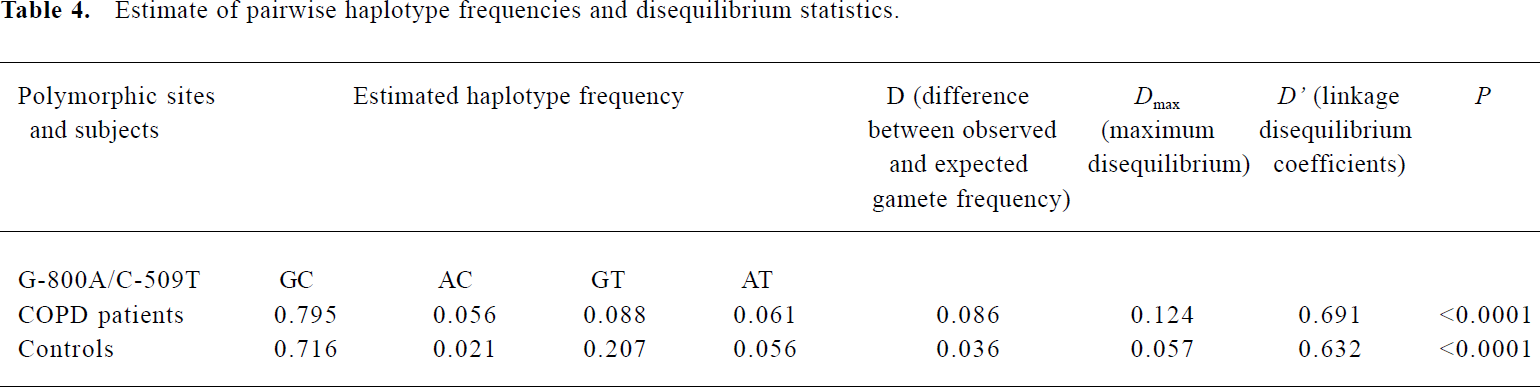
Full table
Discussion
We have shown that the A allele at position -800 in the promoter region of the TGF-β1 gene is more common and the T allele at position -509 is less common in subjects with COPD than in control subjects. Some evidence has shown a correlation between the 2 polymorphisms (G-800A, C-509T) and the concentration of TGF-β1 in human plasma[22]. The G-800A substitution is thought to disrupt a consensus half-site for the binding of the nuclear transcription factor CRE (cyclic AMP-responsive element)-binding protein, and it was found that the presence of the A allele is significantly associated (P<0.05) with lower levels of total TGF-β1 in the circulation[22]. Conversely, the -509T allele has been shown to be associated with higher levels of TGF-β1 in serum and increased TGF-β1 mRNA in peripheral blood mononuclear cells[22]. These findings indicate that the G-800A and C-509T polymorphisms may be involved in the modulation of expression of the TGF-β1 gene and therefore a predisposition for COPD could be linked to particular alleles of this gene. The high producer genotype for TGF-β1 may protect against the development of COPD. A recent study by Wu et al has shown that the +869Pro allele, associated with higher serum concentrations of TGF-β1, was less common in subjects with COPD than in control subjects[16].
A recent study using an animal model of emphysema adds plausibility to the suggestion that increased production of TGF-b1 can protect against the development of COPD. The integrin αvβ6 activates latent TGF-β[23]. Mice that lack this integrin (Itgb6 null mice) develop age-related emphysema. When transgenes for TGF-β1 were inserted into these mice so that they constitutively expressed active TGF-β1, the changes in the lung associated with the deletion of the integrin gene no longer occurred. In these mice, the productive effects of TGF-β1 appear to be related to inhibition of macrophage metalloelastase (MMP12), which degrades elastin.
More than 100 SNP and other genetic variants have been identified in genes of the TGF-β1 signaling pathway, and a few of these have been associated with disease[13]. C-509T was chosen as the sole candidate SNP because previous studies suggest that it is associated with altered serum levels of TGF-β1, asthma diagnosis, asthma severity, and serum IgE levels[22,24,25]. For example, in a study involving 84 monozygous and 86 dizygous twins, Grainger et al showed that TGF-β1 levels in plasma were under genetic control (heritability estimate 0.54) with the C-509T SNP being responsible for 8.2% of the additive genetic variance[22]. The T allele was associated with higher levels of TGF-β1 than the C allele was, and there was an allele dose effect, with the highest levels in TT individuals (7.62 ng/mL), intermediate levels in CT individuals (5.06 ng/mL), and the lowest levels in CC individuals (3.83 ng/mL)[25].
A wide variety of functions have been attributed to TGF-β1 (such as reduction of inflammation and promotion of wound healing[26], immunosuppression[27], regulation of cell proliferation, differentiation and migration[9]), and regulation of extracellular matrix production[28,29]. Some of these functions could protect against the development of COPD. TGF-β1 can inhibit matrix metallopro-teinases that may contribute to the development of emphysema through the digestion of elastic fibers[30,31]. It also promotes the formation of elas-tin[32,33] and this could help repair damage to the lungs of individuals who are smokers and who are risk of developing COPD. Recently, TGF-β1 was located on chromosome 19q, a genomic region linked to the diagnosis of COPD in some genome-wide scans[34]. Our study strengthens this association between TGF-β1 and COPD.
In summary, our results suggest that genetic variation at the promoter of the TGF-β1 gene might be associated with TGF-β1 levels and predisposition to the complex COPD. The distributions of TGF-β1 polymorphisms are different between different ethnic groups. Further replication is essential for the validation of this association in Caucasian, African, or other ethnic populations, and, to elucidate the role of TGF-β1 in COPD, future work is needed to establish the exact effect of TGF-β1 polymorphisms on the activation process of the protein and its function.
References
- Davis RM, Novotny TE. The epidemiology of cigarette smoking and its impact on chronic obstructive pulmonary disease. Am Rev Respir Dis 1989;140:S82-S84.
- Molfino NA. Impact of basic research on tomorrow’s medicine: genetics of COPD. Chest 2004;125:1929-40.
- Kauffmann F, Tager IB, Munoz A, Speizer FE. Familial factors related to lung function in children aged 6–10 years. Results from the PAARC epidemiologic study. Am J Epidemiol 1989;129:1289-99.
- Poller W, Meison C, Olek K. DNA polymorphisms of the α1-antitrypsin gene region in patients with chronic obstructive pulmonary disease. Eur J Clin Invest 1990;20:1-7.
- Samilchuk EI, Chuchalin AG. Mis-sense mutation of α1-antichymotrypsin gene and chronic lung disease Lancet 1993;342:624. [letter].
- Smith CAD, Harrison DJ. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet 1997;350:630-33.
- Schellenberg D, Pare PD, Weir TD, Spinelli JJ, Walker BAM, Sandford AJ. Vitamin D binding protein variants and the risk of COPD. Am J Respir Crit Care Med 1998;157:957-61.
- Huang SL, Su CH, Chang SC. Tumor necrosis factor-agene polymorphism in chronic bronchitis. Am J Respir Crit Care Med 1997;156:1436-9.
- Massague J. The transforming growth factor-β family. Annu Rev Cell Biol 1990;6:597-641.
- Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J Cell Biol 1990;110:1361-7.
- Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast conditioned medium. J Cell Biol 1988;106:1659-65.
- Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, et al. Human transforming growth factor-β complementary DNA sequence and expression in normal and transformed cells. Nature 1985;316:701-5.
- Watanabe Y, Kinoshita A, Yamada T, Ohta T, Kishino T, Matsumoto N, et al. A catalog of 106 single-nucleotide polymorphisms (SNPs) and 11 other types of variations in genes for transforming growth factor-β1 (TGF-β1) and its signaling pathway. J Hum Genet 2002;47:478-83.
- Cambien F, Ricard S, Troesch A, Mallet C, Generenaz L, Evans A, et al. Polymorphisms of the TGF-β1 gene in relation to myocardial infarction and blood pressure. The Etude Cas-Temoin de l’Infarctus du Myocarde (ECTIM) Study. Hypertension 1996;28:881-7.
- Syrris P, Carter ND, Metcalfe JC, Kemp PR, Grainger DJ, Kaski JC, et al. Transforming growth factor-β1 gene polymorphisms and coronary artery disease. Clin Sci 1998;95:659-67.
- Wu L, Chau J, Young RP, Pokony V, Mills GD, Hopkins R, et al. Transforming growth factor-beta1 genotype and susceptibility to chronic obstructive pulmonary disease. Thorax 2004;59:126-9.
- Silverman ES, Palmer LJ, Subramaniam V, Hallock A, Mathew S, Vallone J, et al. Transforming growth factor-β1 promoter polymorphism C-509T is associated with asthma. Am J Respir Crit Care Med 2004;169:214-19.
- Miller SA, Dykes DD, Polesky HF. A simple salting-out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215.
- Xie X, Ott J. Testing linkage disequilibrium between a disease gene and marker loci. Am J Hum Genet 1993;53:1107.
- Zhao JH, Curtis D, Sham PC. Model-free analysis and permutation test for allelic associations. Hum Hered 2000;50:133-39.
- Zhao JH. 2LD- two-locus linkage disequilibrium (LD) calculator. [cited 2002] Available from: http://www.iop.kcl.ac.uk/IoP/Departments/PsychMed/GEpiBSt/software.shtml
- Grainger DJ, Heathcote K, Chiano M, Snieder H, Kemp PR, Metcalfe JC, et al. Genetic control of the circulating concentration of transforming growth factor type β1. Hum Mol Genet 1999;8:93-7.
- Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, et al. Loss of integrin αvβ6 mediated TGF-β activation causes MMP12-dependent emphysema. Nature 2003;422:169-73.
- Hobbs K, Negri J, Klinnert M, Rosenwasser LJ, Borish L. Interleukin-10 and transforming growth factor-β promoter polymorphisms in allergies and asthma. Am J Respir Crit Care Med 1998;158:1958-62.
- Pulleyn LJ, Newton R, Adcock IM, Barnes PJ. TGFβ1 allele association with asthma severity. Hum Genet 2001;109:623-7.
- Pratsinis H, Giannouli CC, Zervolea I, Psarras S, Stathakos D, Kletsas D. Differential proliferative response of fetal and adult human skin fibroblasts to transforming growth factor-beta. Wound Repair Regen 2004;12:374-83.
- Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 2001;29:117-29.
- Ravanti L, Häkkinen L, Larjava H, Saarialho-Kere U, Foschi M, Han J, . Transforming growth factor-beta induces collagenase-3 expression by human gingival fibroblasts via p38 mitogen-activated protein kinase J Biol Chem 1999; 274: 37 292–300.
- Leivonen SK, Chantry A, Häkkinen L, Han J, Kähäri VM. Smad3 mediates transforming growth factor-beta-induced collagenase-3 (matrix metalloproteinase-13) expression in human gingival fibroblasts. J Biol Chem 2002; 277: 46 338–46.
- Eickelberg O, Kohler E, Reichenberger F, Bertschin S, Woodtli T, Erne P, et al. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-beta3. Am J Physiol 1999;276:L814-24.
- Fang KC, Wolters PJ, Steinhoff M, Bidgol A, Blount JL, Caughey GH. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-beta. J Immunol 1999;162:5528-35.
- McGowan SE, Jackson SK, Olson PJ, Parekh T, Gold LI. Exogenous and endogenous transforming growth factors-beta influence elastin gene expression in cultured lung fibroblasts. Am J Respir Cell Mol Biol 1997;17:25-35.
- Kucich U, Rosenbloom JC, Abrams WR, Rosenbloom J. Transforming growth factor-beta stabilizes elastin mRNA by a pathway requiring active Smads, protein kinase C-delta, and p38. Am J Respir Cell Mol Biol 2002;26:183-8.
- Celedon JC, Lange C, Raby BA, Litonjua AA, Palmer LJ, DeMeo DL, et al. The transforming growth factor-beta1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD). Hum Mol Genet 2004;13:1649-56.