
Thrombin-like enzymes from venom gland of Deinagkistrodon acutus: cDNA cloning, mechanism of diversity and phylogenetic tree construction1
Introduction
Snake venom is a rich source of substances with therapeutic value, and its components vary between species. Some proteins and peptides in snake venom have enormous structural diversity and they are thought to be genetically determined by multiple gene families. This feature of structural diversity confers on snakes certain adaptive advantages during their evolution. Collateral evidence for this is that Aipysurus eydouxii, a sea snake that feeds exclusively on fish eggs, possesses atrophied venom glands and has PLA2 toxins with less diversification[1]. Similar variation has also been observed in other venomous animals, such as scorpions[2].
Snake venom thrombin-like enzymes (TLE) are mainly distributed in Viperidae snakes, and are especially abundant in Deinagkistrodon acutus. Anti-thrombotic agents have been developed from TLEs of snake venom and are in clinical use. Both the TLE proteins and mRNAs so far isolated display accelerated evolution, that is, their structures are remarkably diversified. Actually, while cloning snake venom TLE cDNAs, it was found to be practically impossible to find 2 totally identical cDNA molecules[3]. Under such circumstances, it seems unlikely that each cDNA would correspond to a specific gene in the genome. The diversity is generally thought to be attributable to such mechanisms as gene mutation, gene conversion or alternative splicing of pre-mRNA[4]. However, no definitive conclusion can yet be drawn regarding the molecular mechanism by which their structural diversity is achieved. Therefore, research effort in this area may not only help to reveal the molecular evolution of snake venom proteins, but also improve our understanding of their structure-function relationships and provide guidance for better exploitation of these substances, especially with respect to recombinant production.
In the present study, some new TLE cDNAs from Deinagkistrodon acutus were cloned. The cDNA sequences were aligned and analyzed to determine possible rules for sequence variations. A phylogenetic tree was constructed and discussed.
Materials and methods
Materials Kits for RNA extraction, reverse transcription, and plasmid rapid extraction were purchased from BioDev-Tech Scientific and Technical Co, Beijing, China. Taq DNA polymerase and T4 DNA ligase were from Beijing Sino-American Company. The competent Escherichia coli cell preparation kit was the product of Sangon, Shanghai, China. Plasmid pGEM-T and E coli JM109 strain were from Promega. Four adult Deinagkistrodon acutus snakes were collected from Hunan Province, China.
Reverse transcription-polymerase chain reaction and cDNA cloning Isolation of the total RNA from a snake venom gland and the reverse transcription-polymerase chain reactions (RT-PCR) were performed as described previously[3]. The total RNA in the experiment was a preparation from a single snake venom gland. For PCR, a pair of degenerate primers was designed on the basis of the highly-conserved N- and C-termini amino acid sequences of TLEs: T1 5'-GTC ATT GGA GGT GA(TC) GA(AG) T-3'; T2 5'-A(CT)G GGG GGC AAG T(TC)G C(AG)-3'. The 5' dATP was designed as per the first nucleotide in the stop codon.
The PCR products amplified with Taq DNA polymerase were cloned into the pGEM-T vector and the inserted cDNAs were sequenced with ABI Prism 377-96 by Sangon. Each cDNA was sequenced in two directions.
Software cDNAs were translated and exported with VISED, a freely distributed software (downloaded from http://iubio.bio.indiana.edu). Sequence alignment was performed with Omiga 2.0 (developed by Oxford Molecular Office)[5] and exported with GenDoc (downloaded from http://www.cris.com). Phylogenetic trees were constructed using Megalign[6]. Similarity searches were carried out online using the BLAST program at the website http://www.ncbi.nlm.nih.gov.
Results
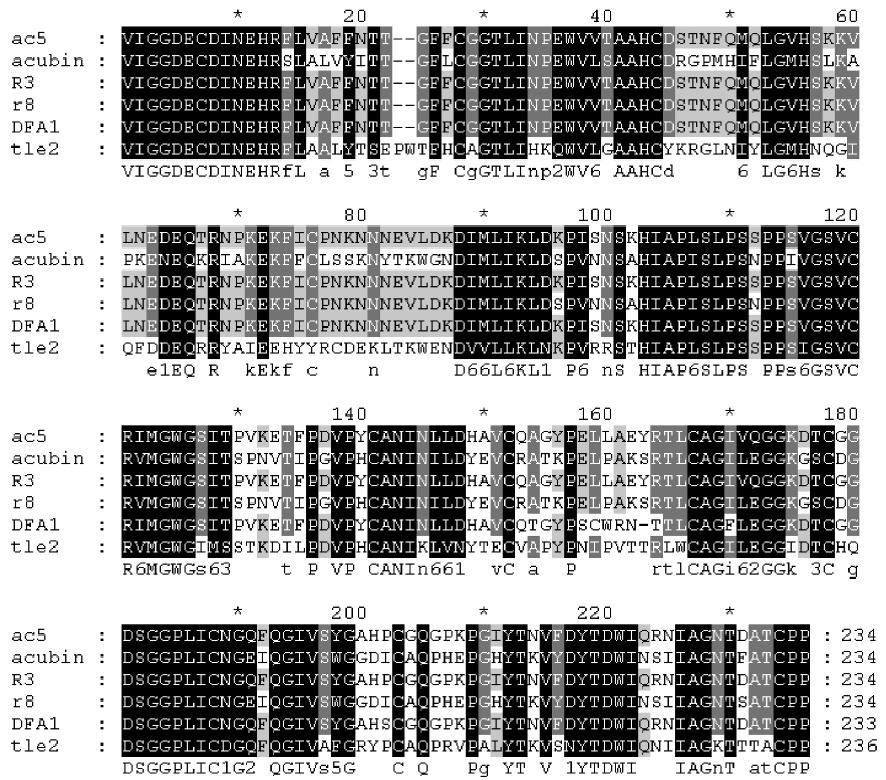
By using the RT-PCR and cDNA cloning strategy, we obtained 7 complete cDNA sequences coding for mature TLEs, that is, ac1, ac5, ctq, n1, R3, R5, and tler7. We compared the cDNA sequences (Figure 1), and the statistical reports show that the sequence identities range from 81% to 99%. Despite the differences, the TLE sequences have certain common features. First, all the cloned cDNAs have open reading frames that encode 234 amino acids. Second, their deduced amino acid sequences were homologous with known snake venom TLEs, and an NCBI Conserved Domain Search[7] demonstrated that they were all homologous with trypsin-like serine protease domains (CD accession numbers cd00190, smart00020, pfam00089, and COG5640). Third, no amino acid substitution of the catalytic triad residues was found, which suggests that the proteins were probably functionally active enzymes.

ctq By using the BLAST program we demonstrated that the amino acid sequence deduced from ctq cDNA had 99% identity with the thrombin-like enzyme precursor (GenPept accession number AAK12273) and 83% identity with acubin (GenPept accession number CAB46431) from Deinagkistro-don acutus.
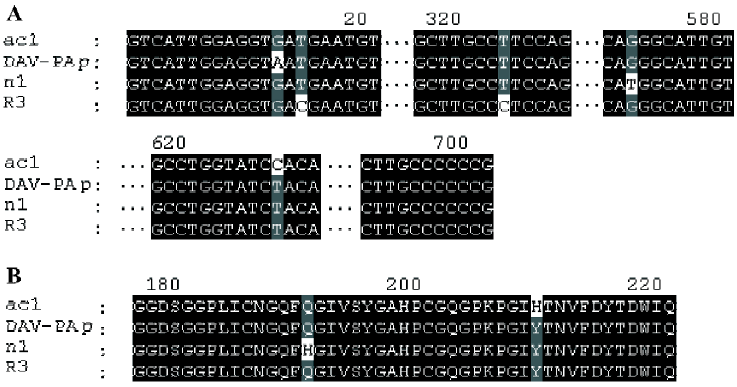
ac1, ac5, R3, and n1 On the basis of BLAST analysis, the amino acid sequence of ac1, ac5, R3, and n1 had the highest degree of identity (99%) with the DAV-PA pre-cursor[8] (GenPept accession number AAF76378). Points of difference from the DAV-PA precursor are that these sequences have Asp-5 instead of Asn-5, which is a conservative substitution; furthermore, Gln-191 is replaced by His-191 in n1, and His-210 substitutes for Tyr-210 in ac1. Comparison of the cDNAs of R3 and the DAV-PA precursor indicates that there is a synonymous base substitution of CCT with CCC, which both encode Ser-108.
ac1, ac5, R3, and n1 had the second highest degree of identity (94%) with DFA1 (Deinagkistrodon acutus thrombin-like defibrase1, GenPept accession number AAD19350)[9]. DFA1 has only 233 amino acid residues because it lacks Tyr-161 of ac1, ac5, R3 and n1.
R5 and tler7 R5 cDNA has only a single base difference from the cDNA of the thrombin-like enzyme precursor of Deinagkistrodon acutus (GenBank accession number AF333768); that is, at site 612 the former has an A and the latter has a G. But this change occurs at the third position of the codon, and both CCA and CCG code for proline. There-fore, the amino acid sequences remain unchanged.
tler7 also had the highest degree of identity (85%) with thrombin-like enzyme precursor (AAK12273), and its degree of identity with acubin was 83%. tler7 cDNA was accepted by GenBank, and its accession number is AF362127.
Abnormal sequence T4 An abnormal sequence, T4, was also cloned. Although T4 cDNA is similar to the other cDNAs, it has 681 bp (shorter than the usual 702 bp), and it would encode a truncated peptide, because a nonsense codon appears in the middle of the reading frame. We compared the sequence with those of other TLEs and with the batroxobin gene (GenBank accession number X12747), a thrombin-like enzyme from Bothrops atrox moojeni venom[10], and found that a segment of T4, GFPLNGFERQYFLFQAMRSA-PLVGDNGNYSSMHLGGKLZ was aberrant. It replaced the segment normally encoded by exon 3, with the batroxobin gene as reference, and BLAST analysis of its cDNA showed that it has 91% identity with a region of intron 3 that conforms to the Breathnach-Chambon rule[11]. Intron 3 has a tandem repeat of TTGGTTGGAGACAATGGAAA (from 6712 to 6751) in the region (Figure 2). These results suggest three things. First, the gene structures of TLEs might be similar in Deinagkistrodon acutus and Bothrops atrox. Second, a putative minisatellite site exists in intron 3. Third, T4 is a possible product of alternative splicing.

Discussion
Factors contributing to variations Snake venom TLEs exhibit great variability, but their molecular scaffolding is conserved[12]. From a genetic viewpoint, conservativeness and variability are normally maintained in a delicate balance that is probably ensured by a set of mechanisms or a “super-visor”. As for the TLEs, the balance seems to incline towards variability. TLEs resemble antibodies in that they also could be seen as armaments against all kinds of exterior or environmental factors. A relaxed mechanism is used and the possibility of variation is enhanced. Evolution has selected the changeability itself.
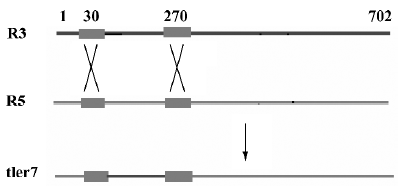
Multiple alignments suggested that the variation could be categorized into three types: type I, II, and III. Type I relates to the differences involved in relatively large seg-ments. The alignment of the cDNAs of R3, R5 and tler7 illustrates variation of this type. Continuous sequence identities between R3 and tler7, and between R5 and tler7, suggest that the combination of R3 and R5 produced tler7 (Figure 3). Judging by the organization of the batroxobin gene, so far the only known genomic structure of TLEs of snake venom, it is obvious that the combination is neither the result of alternative splicing of the pre-mRNAs nor that of trans-splicing. A similar phenomenon was reported by Siigur et al[13]. We propose that the variation is in fact the result of post-transcriptional recombination, as we have previously hypothesized for snake venom C-type lectin proteins[14,15]. The putative crossover sites at around 20–40 and 260–280 were shared by the 3 cDNAs (Figure 4), and some other dispersed sequence identities in R3, R5 and tler7 were also observed. At the present time it is not clear whether these regions are a prerequisite for or a consequence of recombination. If the former were true, the conserved crossover sites could be explained as the feature of the gene family; whereas if the latter were true we would infer that the recombination was proceeding continually in the live venom gland.

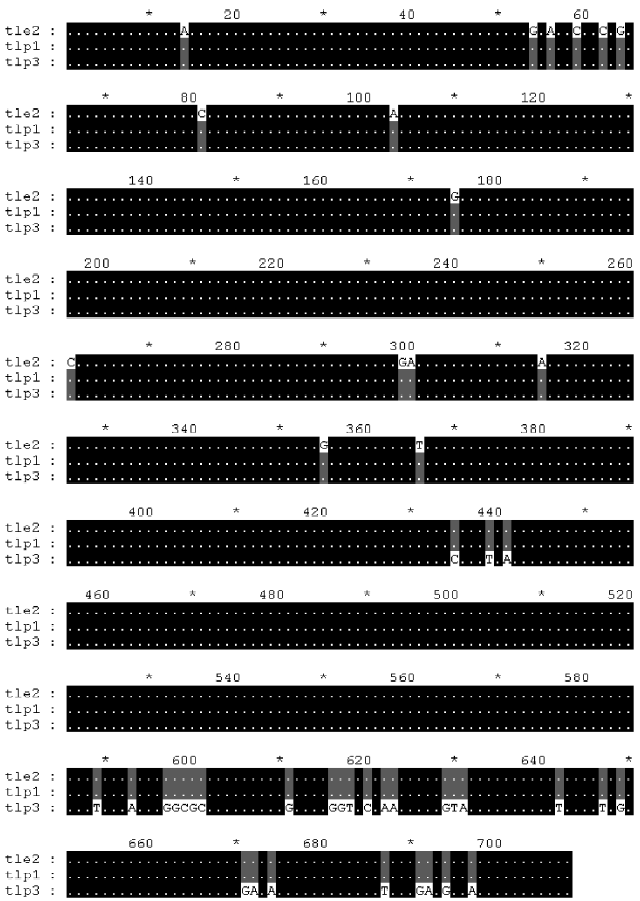
Furthermore, we analyzed 3 cDNA sequences from GenBank: Deinagkistrodon acutus thrombin-like protein 1 (AY861382), thrombin-like enzyme 2 (AY861138) and thrombin-like protein 3 (AY861383). Multiple alignment of the 3 sequences provided additional evidence for the recombination hypothesis (Figure 5).
The characteristics of the recombination coincide with those of some homologous or non-replicative homologous RNA recombination models[16]. The mechanism and the enzymatic basis of this proposed post-transcriptional recombination remain to be elucidated, and its evolutionary origin is also mysterious. Viral RNA genomes undergo rapid evolution[17], and so far RNA recombination has been found only in RNA molecules that have genomic functions, such as genomes of RNA viruses, excluding RNA processing[18,19]. We wonder if there exist evolutionary connections between eukaryotic recombination and viral genomic recombination.
Type II variation in alignment relates to point mutations, including deletions/insertions of one or several bases and base substitutions (missense mutations or synonymous/conservative mutations) as illustrated in Figures 6 and 7. For base substitutions, all 6 possible shifts have been found, but shifts between A and G seem to occur with the greatest frequency, according to our preliminary statistics. In some alignments, shifts between C and T were the second most frequent. Our BLAST analyses and comparison data suggest that type II variations were the most widely dispersed in the present study. The origins of type II variations are not yet understood, but RNA editing, gene conversion or point mutation caused by RNA recombination might be the primary cause[20,21]. Therefore, the relationships between type I and II variations are worth studying in the future.
Type III variation in alignment comprises mistakes. T4 is not the sole example of this type. We have also cloned another partial cDNA sequence, O1, which had the same abnormal splicing as T4 cDNA; that is, a segment of intron 3 took the place of exon 3. However, O1 is different from T4 cDNA because of some type II variations. These findings again highlight the instability inherent in the flow of genetic information in snake venom.
The preceding discussion is based on data gathered using some traditional techniques, such as PCR. Therefore, replication errors or template switching in DNA amplification should be considered. To circumvent these potential problems, high-fidelity Pfu DNA polymerase was also used to validate the accuracy of PCR reactions catalyzed by Taq DNA polymerase, and the errors proved to be negligible as far as our experiments were concerned. In future studies, cDNA libraries and genomic DNA libraries should be used. However, an overall understanding of snake genomes is necessary to answer questions regarding the diversity of TLEs.
Horizontal gene transfer Alignment of defirase 1, acubin and ancrod (Figure 8) revealed a segment that is peculiar to ancrod: PRTRWGE (81–87)[22]. Ancrod is purified from the venom of Calloselasma rhodostoma, and is being clinically used for the treatment of conditions such as acute ischemic stroke. BLAST analysis (expect=10000) of the segment hit only 2 unknown proteins from Arabidopsis thaliana (aside from ancrod itself), and the degree of similarity between them was 85%. That segment probably represents a novel motif, and it possibly originated from other organisms through horizontal gene transfer. A retrovirus might have played a role in the horizontal transfer[14,23].

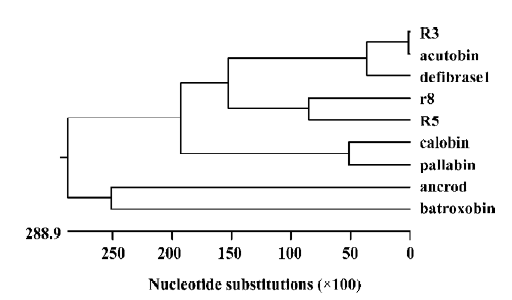
Phylogenetic analysis Phylogenetic trees (Figure 9) constructed by using some TLE amino acid sequences are consistent with conventional taxonomy; that is, the degree of variation is greater between species than within species. TLEs seem to have some value for relationship estimation. However, TLEs of the species Deinagkistrodon acutus do not possess geographic features according to our phylogenetic analysis. Wang et al constructed TLE phylogenetic trees to find functional groups[8]. Because functional alterations may involve a very limited number of key amino acid residues, direct experiments on structure-function relationships at the DNA and protein level need to be conducted: an important task for studying the structural biology of TLEs.
References
- Li M, Fry BG, Kini RM. Putting the brakes on snake venom evolution: the unique molecular evolutionary patterns of Aipysurus eydouxii (marbled sea snake) phospholipase A2 toxins. Mol Biol Evol 2005;22:934-41.
- Srinivasan KN, Sivaraja V, Huys I, Sasaki T, Cheng B, Kumar TKS, et al. κ-Hefutoxin1, a novel toxin from the scorpion Heterometrus fulvipes with unique structure and function. J Biol Chem 2002;277:30040-7.
- Zha XD, Ren B, Liu J, Xu KS. cDNA cloning and high-level expression of a thrombin-like enzyme from Agkistrodon acutus venom. Methods Find Exp Clin Pharmacol 2003;25:253-7.
- Deshimaru M, Ogawa T, Nagashima K, Nobuhisa I, Chijiwa T, Shimohigashi Y, et al. Accelerated evolution of crotalinae snake venom gland serine proteases. FEBS Lett 1996;397:83-8.
- Searby C. OMIGA 2.0. Biotech Software Internet Rep 2000;1:198-207.
- Clewley JP, Arnold C. MEGALIGN. The multiple alignment module of LASERGENE. Methods Mol Biol 1997;70:119-29.
- Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res 2004;32:W327–31.
- Wang YM, Wang SR, Tsai IH. Serine protease isoforms of Deinagkistrodon acutus venom: cloning, sequencing and phylogenetic analysis. Biochem J 2001;354:161-8.
- Pan H, Du X, Yang G, Zhou Y, Wu X. cDNA cloning and expression of acutin, a thrombin-like enzyme from Agkistrodon acutus. Biochem Biophys Res Commun 1999;255:412-5.
- Itoh N, Tanaka N, Funakoshi I, Kawasaki T, Mihashi S, Yamashina I. Organization of the gene for batroxobin, a thrombin-like snake venom enzyme. Homology with the trypsin/kallikrein gene family. J Biol Chem 1988;263:7628-31.
- Breathnach R, Benoist C, O’hare K, Gannon F, Chambon P. Ovalbumin gene: evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc Natl Acad Sci USA 1978;75:4853-7.
- Fry BG, Wüster W. Assembling an arsenal: origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences. Mol Biol Evol 2004;21:870-83.
- Siigur E, Aaspollu A, Siigur J. Sequence diversity of Vipea lebetina snake venom gland serine protease homologs: result of alternative-splicing or genome alteration. Gene 2001;263:199-203.
- Zha XD, Zhou LZ, Huang HS, Liu J, Xu KS. Analysis of cDNAs and genomic DNA of snake venom CTL-like proteins revealed an extraordinary post-transcriptional processing event. Chin J Biochem Mol Biol 2004;20:713-8.
- Zha XD, Liu J, Xu KS. cDNA cloning, sequence analysis and recombinant expression of akitonin beta, a C-type lectin-like protein from Agkistrodon acutus. Acta Phamacol Sin 2004;25:372-7.
- Onodera S, Sun Y, Mindich L. Reverse genetics and recombination in Φ8, a dsRNA bacteriophage. Virology 2001;286:113-8.
- Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, Van de Pol S. Rapid evolution of RNA genomes. Science 1982;215:1577-85.
- Alejska M, Kurzyñska-Kokorniak A, Broda M, Kierzek R, Figlerowicz M. How RNA viruses exchange their genetic material. Acta Biochim Pol 2001;48:391-407.
- Nagy PD, Zhang CX, Simon AE. Dissecting RNA recombination in vitro: role of RNA sequences and the viral replicase. EMBO J 1998;17:2392-403.
- Blanc V, Davidson NO. C-to-U RNA editing: mechanisms leading to genetic diversity. J Biol Chem 2003;278:1395-8.
- Maas S, Rich A, Nishikura K. A-to-I RNA editing: recent news and residual mysteries. J Biol Chem 2003;278:1391-4.
- Au LC, Lin SB, Chou JS, Teh GW, Chang KJ, Shih CM. Molecular cloning and sequence analysis of the cDNA for ancrod, a thrombin-like enzyme from the venom of Calloselasma rhodostoma. Biochem J 1993;294:387-90.
- Martin J, Herniou E, Cook J, O’Neill RW, Tristem M. Interclass transmission and phyletic host tracking in murine leukemia virus-related retroviruses. J Virol 1999;73:2442-9.