
Hydrogen sulfide ameliorates vascular calcification induced by vitamin D3 plus nicotine in rats1
Introduction
Vascular calcification is a common finding in many cardiovascular diseases, such as hypertension, atherosclerosis, diabetes, chronic renal failure, aging, and arterial stenosis, and is also common after prosthetic valve replacement[1–3]. Calcified vessels have decreased capacity for vasodilatation, and increased stiffness, and promote a form of thrombus and atherosclerotic plaque rupture. Vascular calcification is now recognized as a marker of atherosclerotic plaque burden, as well as a major contributor to the loss of arterial compliance and increased pulse pressure that is seen with aging, diabetes, and renal insufficiency. It is an important risk factor for cardiovascular disease[4,5]. However, the mechanism by which vascular calcification causes vascular dysfunction and remodeling is unclear[6,7].
Previously, calcification was generally considered to be a process of passive calcium deposition in the extracellular matrix and cells. That view, however, has changed in recent years, and vascular calcification is now considered to be an active, regulative process similar to osteogenesis[8]. During vascular calcification, the various vascular cells, including vascular smooth muscle cells, pericytes, fibroblasts and macrophages, transform into an osteoblast-like phenotype, which is characterized by increased alkaline phosphatase (ALP) activity, matrix vesicle formation and overexpression of bone morphogenetic proteins (BMP) including BMP-2 and bone matrix proteins such as osteopontin (OPN), osteonectin and osteocalcin[5,9–11].
However, changes in the function of vascular cells with the phenotypic alteration and the pathophysiological significance of the altered phenotype remain unclear. It is common knowledge that paracrine/autocrine factors secreted from vascular cells contribute to circulatory homeostasis and mediate the pathogenesis of cardiovascular diseases. Recent research has shown that paracrine/autocrine dysfunction in calcified vascular vessels plays an important role in calcification-induced vascular damage. Vasoactive substances produced by cardiovascular tissues including adrenomedullin[12,13], endothelin[14], C-type natriuretic peptide, parathyroid hormone-related peptide[15], growth factor[16], cytokine[17] and the gaseous transmitters nitric oxide (NO) and carbon monoxide (CO) are involved in the pathophysiological process of vascular calcification[18,19].
It is well known that gaseous transmitters such as NO and CO participate in the regulation of the pathophysiological process of cardiovascular disease, an important target of therapeutic drugs for cardiovascular disease. Our previous research showed that the L-arginine/NO synthase/NO/cGMP and heme/heme oxygenase/CO/cGMP pathways were altered during vascular calcification, which suggests that NO and CO play important roles in the pathogenesis of vascular calcification[18,19]. Endogenous hydrogen sulfide (H2S) is a newly discovered gaseous transmitter. Two pyridoxal-5-phosphate-dependent enzymes, cystathionine β-synthase (CBS; EC 4.2.1.22) and cystathionine γ-lyase (CSE; EC 4.4.1.1), are responsible for most of the endogenous production of H2S in mammalian tissues that use L-cysteine as the main substrate[20]. Cardiovascular tissues rich in CSE, which are an important source of endogenous H2S and have been shown to have vasodilatory, hypotensive, and negative inotropic and growth-regulating properties, contribute to vascular homeostasis[21–25]. However, the pathophysiological significance of the endogenous CSE/H2S pathway in vascular calcification is unclear. In this work, we developed a rat vascular calcification model induced by vitamin D3 plus nicotine (VDN) to observe alterations in the vascular CSE/H2S pathway and the effects of treatment with H2S on vascular calcification, to explore the significance of endogenous H2S in the pathogenesis of vascular calcification.
Materials and methods
Materials All animal care and experimental protocols complied with the animal management guidelines of the Chinese Ministry of Health (document 55, 2001) and the Animal Care Committee of the First Hospital, Peking University. Male Sprague-Dawley rats (weight 212±2 g) were obtained from the Animal Center, Health Science Center, Peking University (Beijing, China). NaHS, L-cysteine, pyridoxal-5'-phosphate, and vitamin D3 plus nicotine were purchased from Sigma (St Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM) and Trizol were obtained from Gibco (Rockville, MD, USA). 45CaCl2 was obtained from NEN Life Science (Boston, MA, USA). Specific primers for the amplification of OPN were: sense (OPN-S) 5'-CTC GCG GTG AAA GTG GCT GA-3', and antisense (OPN-A) 5'-GAC CTC AGA AGA TGA ACT CT-3'. Primers for the amplification of CSE were: sense (CSE-S) 5'-TCC GGA TGG AGA AAC ACT TC-3', and anti-sense (CSE-A) 5'-GCT GCC TTT AAA GCT TGA CC-3'; and those for the amplification of β-actin (for calibrating sample loading) were: sense (β-actin-S) 5'-ATC TGG CAC CAC ACC TTC-3', and antisense (β-actin-A) 5'-AGC CAG GTC CAG ACG CA-3'. These oligonucleotide primers were synthesized by SBS (Sai Bai Sheng, Beijing, China). Other chemicals and reagents were of analytical grade.
Preparation of rat vascular calcification model We used a model version of the protocol originally described by Niederhoffer et al[26]. Rats in the VDN group received vitamin D3 (300 000 IU/kg, im) and nicotine (25 mg/kg, orally) at 9:00 on d 1. Nicotine administration was repeated at 19:00. Rats in the control group received an injection of normal saline (im) and two gavages of vehicle. The rats treated with VDN were intraperitoneally injected with 2.8 or 14 μmol/kg per d of freshly prepared NaHS (H2S donor) for 4 weeks, corresponding to low-dose and high-dose NaHS groups, respectively. All rats were housed under standard conditions (room temperature 20±1 °C, humidity 60%±10%, light from 6:00 to 18:00) and given standard rodent chow and water freely.
At the end of week 4, the rats were anesthetized with sodium pentobarbital (45 mg/kg, ip), and catheters filled with heparin saline (500 U/mL) were inserted into the right femoral and right carotid arteries for measuring arterial pressure and intraventricular pressure, respectively. A blood sample was drawn and mixed with 1 mg/mL ethylenediamine tetraacetic acid-Na2 and 500 KIU/mL of aprotinin. Plasma was obtained by centrifugation at 1600×g for 15 min at 4 °C and stored at -70 °C. The intact aortas were harvested, weighed and stored at -70 °C until use.
Measurement of calcium content in aorta Calcium content in the aorta was determined as described previously[13]. The aortas (~10 mg) were dissolved in HNO3 and diluted with a blank solution (27 nmol/L KCl, 27 μmol/L LaCl3). The calcium content was measured on an atomic absorption spectrophotometer at 422.7 nm (novAA 300; Analytik Jena AG, Germany).
Calcification assay (45Ca accumulation) As described in a previous paper[13], aortic tissue (~20 mg) was sliced and incubated in 1 mL Krebs-Henseleit (K-H) solution (in mmol/L: 118 NaCl, 4.7 KCl, 1.3 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, and 5 glucose; pH 7.2) with 37 kBq/mL of 45CaCl2. The reaction was stopped by the addition of ice-cold K-H solution. The tissue was dissolved and protein content was determined by using Bradford’s method[4]. 45Ca2+ radioactivity was measured by β-scintillation counting (LS 6500; Beckman).
Measurement of ALP activity in aortas Tissue ALP activity was measured as described previously[13]. An aortic homogenate was prepared in homogenizing buffer [20 mmol/L N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), pH 7.4, containing 0.2% NP-40, and 20 mmol/L MgCl2] with a Polytron (Tekmar Company, Germany) homo-genizer. After centrifugation at 8000×g for 10 min, the supernatant was collected. The protein content of the tissue supernatant was determined by Bradford’s method[4]. An ALP activity assay was performed by mixing 200 µg of protein sample (in 200 µL) with 1 mL reaction mixture (alkaline buffer:stock substrate solution, 1:1) with a modification of the ALP assay kit from Sigma. This mixture was then incubated for 30 min at 37 °C. Yellow color was indicative of ALP activity. The reaction was stopped by the addition of 12 µL of 1 mol/L NaOH, and absorbance was determined at 405 nm. ALP activity was calculated with ρ-nitrophenol used as a standard, according to the manufacturer of the kit’s instructions (Sigma). One unit was defined as the activity producing l nmol of ρ-nitrophenol for 30 min.
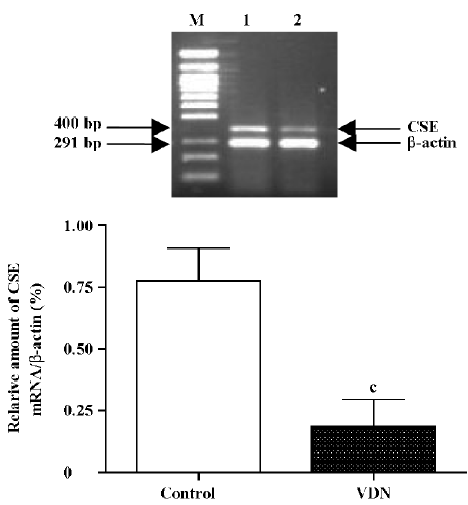
Measurement of OPN and CSE mRNA in aortas The concentration of OPN and CSE mRNA was determined by reverse transcription-polymerase chain reaction (RT-PCR)[27]. Total aortic RNA was prepared by in situ lysis with Trizol reagent. One microgram of total tissue RNA was reverse-transcribed into single strand cDNA with M-MuLV reverse transcriptase and oligo (dT) 15 primers. PCR was performed in a 0.2 mL tube containing 2 µL tissue cDNA, 5 µmol/L of each of the OPN-S and OPN-A primers (1 µL), 2.5 mmol/L of each dNTP (1 µL), 1.5 mmol/L MgCl2 (1.5 µL), 10× PCR buffer (2.5 µL), and 1.25 units of Taq DNA polymerase, in a total volume of 25 µL. After being denatured at 95 °C for 5 min, the solution underwent PCR at 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 40 s for 28 cycles, and then 72 °C for 5 min. Seven microliters of PCR product was separated on a 1.5% agarose gel and stained with ethidium bromide. The optical density of the 871 bp band was measured by use of the Gel Documentation System (Bio-Rad, Hercules, CA, USA). Amplification of OPN cDNA was confirmed with analysis with an OPN primer measuring ribonucleotide sequence. Two microliters of PCR product was amplified again with the 2 rat β-actin primers at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 40 s for 23 cycles, and then at 72 °C for 5 min, and the optical density of the β-actin band (291 bp) was measured. The relative amount of CSE mRNA (400 bp) was determined according to the method described earlier. The solution underwent PCR at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 40 s for 30 cycles.
Measurement of CSE activity and H2S content in plasma and aortas Tissue CSE activity was measured as described previously[21], with minor modifications. Briefly, aortic tissue homogenate was suspended in 50 mmol/L ice-cold potassium phosphate buffer (pH 6.8). The reaction mixture contained (in mmol/L): 100 potassium phosphate buffer (pH 7.4), 10 L-cysteine, 2 pyridoxal-5'-phosphate, and 10% (w/v) tissue homogenate. Cryovial test tubes (2 mL) were used as the center wells; each contained 0.5 mL of 1% zinc acetate as a trapping solution and a filter paper of 2.0 cm×2.5 cm to increase the air/liquid contact surface. The reaction was performed in a 25 mL Erlenmeyer Pyrex flask. The flasks containing the reaction mixture and the center wells were flushed with N2 before being sealed with a double layer of Parafilm. The reaction was initiated by transferring the flasks from ice to a shaken water bath at 37 °C. After incubation at 37 °C for 90 min, 0.5 mL of 50% trichloroacetic acid was added to stop the reaction. The flasks were sealed again and incubated at 37 °C for another 60 min to ensure complete trapping of the H2S released from the mixture. The contents of the center wells were then transferred to test tubes, each containing 3.5 mL water. Subsequently, 0.5 mL of 20 mmol/L N,N-dimethyl-p-phenylenediamine sulfate in 7.2 mmol/L HCl was added, immediately followed by 0.4 mL of 30 mmol/L FeCl3 in 1.2 mol/L HCl. The absorbance of the resulting solution at 670 nm was measured using a spectrophotometer (DU 640; Beckman). The H2S concentration was calculated using the calibration curve of a standard H2S solution.
The tissue and plasma concentrations of H2S were measured by using the method described earlier, without adding L-cysteine and pyridoxal-5'-phosphate to the reaction mixture. Trichloroacetic acid was added directly into the tissue homogenates and incubated for 60 min; the plasma was then centrifuged and the suspension collected. After adding display fluid to the suspension, optical density was measured at 670 nm. H2S concentration was calculated by using the calibration curve.
von Kossa staining von Kossa staining for aorta was performed according to the method described by Zhang et al[12] . A 1-cm segment of aortic arch was excised and fixed with 10% formalin. Samples were dehydrated and embedded in paraffin. Six-micrometer-thick sections were cut, and some of the slides were stained with hematoxylineosin. Other slides were treated with 5% AgNO3 for 30 min. Specimens were then counterstained with safranine (red staining) and examined under a light microscope.
Statistical analysis The results of aortic ALP activity and 45Ca uptake were normalized to total protein and all data were expressed as mean±SD. For comparisons between 2 groups, the unpaired Student’s t-test was used. One-way ANOVA, followed by the Student-Newman-Keuls test for significance was used to compare the 3 groups. P<0.05 was considered statistically significant.
Results
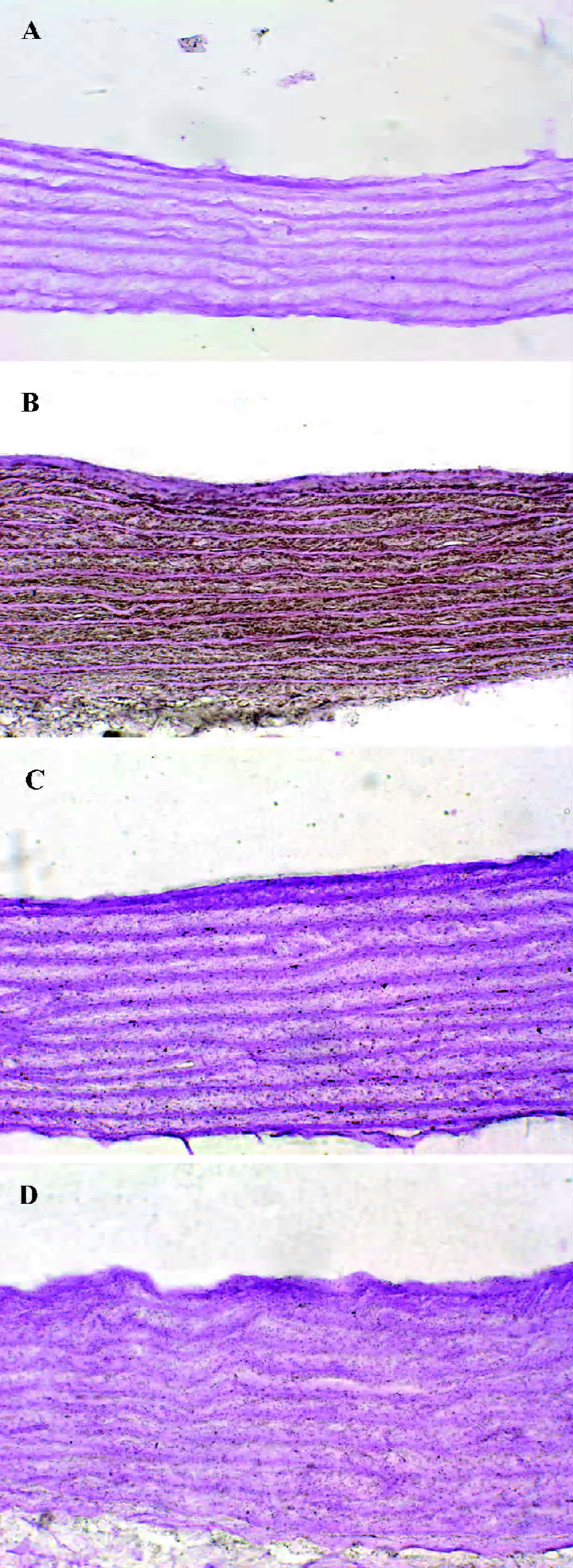
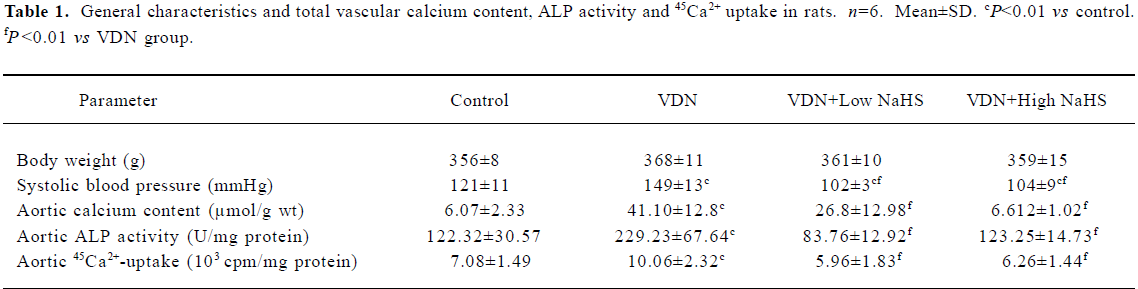
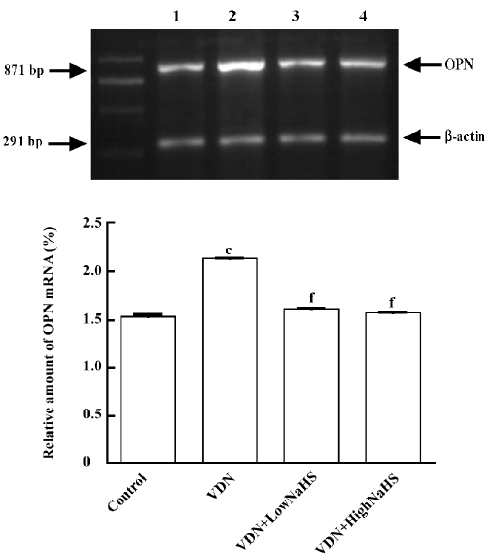
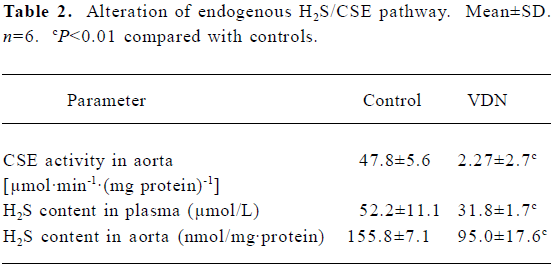
General characteristics of vascular calcification In rats with vascular calcification induced by VDN, systolic blood pressure was 23% higher than the blood pressure of rats in the control group. Von Kossa staining for calcium mineral deposits produced strong positive black/brown-staining areas among the elastic fibers of the medial layer in calcified aorta (Figure 1). Calcium content and 45Ca2+ accumulation in the calcified aorta were significantly increased by 6.8-fold and 1.4-fold, respectively, relative to the control (P<0.01). Aortic ALP activity was significantly increased (by 1.9-fold; P<0.01) relative to the control (Table 1). OPN mRNA concentration was increased by 39% (P< 0.01) relative to the control (Figure 3).
Full table
Downregulation of CSE/H2S pathway in rats with vascular calcification Compared with the controls, the plasma and aorta H2S content was reduced by 39% and 57% (all P<0.01, Table 2) respectively, aortic CSE activity was decreased by 53% (P<0.01,Table 2), and aortic CSE mRNA amount was reduced by 76%, in rats with vascular calcification (P<0.01; Figure 2).
Full table
Administration of NaHS ameliorated vascular calcification Administration of NaHS (a donor of H2S) obviously reduced blood pressure in rats with vascular calcification, compared with rats that did not receive NaHS. Treatment with low and high doses of NaHS reduced systolic blood pressure by 38% (P<0.05) and 30% (P<0.01), respectively (Table1). NaHS significantly reduced the aortic calcium mineral deposits (Figure 1). The rats treated with low doses of NaHS had reduced vascular calcium content, 45Ca2 + accumulation and ALP activity by 35%, 4% and 63% (P<0.01), respectively, and the rats in the high-dose NaHS group had reduced calcium content, 45Ca2 + accumulation and ALP activity by 84%, 38% and 46% (P<0.01), respectively. The levels of aortic OPN mRNA were decreased by 74% (P<0.01) in the low-dose group and by 86% (P<0.01) in the high-dose group, compared with the rats that did not receive NaHS. No significant differences in the above parameters were found between the NaHS-treated groups (P>0.05; Table 1, Figure 3).
Discussion
Pathological calcification of cardiovascular structures, or vascular calcification, is associated with a number of diseases, including end stage renal disease (ESRD), diabetes and cardiovascular diseases. Calcium phosphate deposition, in the form of bioapatite, is the hallmark of vascular calcification, and can occur in the blood vessels, myocardium, and cardiac valves. Calcified deposits are found in distinct layers of the blood vessels and are related to the underlying pathology. Intimal calcification occurs in atherosclerotic lesions[1,2], whereas medial calcification (also known as Monckeberg’s medial sclerosis) is associated with vascular stiffening and the arteriosclerosis observed with aging, diabetes, and ESRD[3]. In coronary arteries, calcification is positively correlated with atherosclerotic plaque burden, increased risk of myocardial infarction, and plaque instability[6,7].
Previously, calcification was generally considered to be a process of passive calcium deposition in the extracellular matrix and cells. However, elevated calcium×phosphorus product (Ca×P) cannot fully explain the pathogenic process of cardiovascular calcification. Growing evidence indicates that vascular calcification is an actively regulated process in which vascular cells are transformed to an osteoblast-like phenotype, which is characterized by an increase in ALP activity, matrix vesicle formation and overexpression of marker proteins of the osteoblast phenotype. Obviously, the disturbance of vascular paracrine/autocrine function is an important pathogenetic cause of vascular calcification. We have reported that adrenomedullin, c-type natriuretic peptides, and parathyroid hormone-related protein secreted from the vasculature can inhibit or delay the pathogenesis of vascular calcification, but endothelin can facilitate or intensify its pathogenesis. Interestingly, NO and CO are protective factors against cardiovascular calcification, and the
H2S is the recently discovered third gaseous signaling molecule. In the central nervous system, endogenous H2S is produced in response to neuronal excitation, and alters hippocampal long-term potentiation (LTP), a synaptic model for memory, increasing the sensitivity of N-methyl-D-aspartate receptors following increased intracellular cAMP, and modulates the hypothalamo-pituitary-adrenal axis function in vitro and in vivo[28]. Cardiovascular tissues are rich in CSE, which catalyzes L-cysteine to generate H2S. H2S has been shown to have vasodilatory, hypotensive, negative inotropic and growth-regulating properties, which contribute to physiological regulation of cardiovascular homeostasis together with NO and CO[18,19]. The endogenous CSE/H2S pathway participates in the pathophysiological process in cardiovascular diseases such as hypoxia-induced pulmonary hypertension[24], spontaneous hypertension[25], NO-deficient hypertension[29], septic and endotoxin shock, and myocardial ischemia[30].
Vitamin D hypervitaminosis can induce an increase in the tissue calcium content, which is deposited mainly on the elastic fibers. Nicotine amplifies the calcifying effects of vitamin D. VDN induces calcium overload in the arteries, media calcification, and finally widespread cardiovascular calcification. In the present study, in rats with vascular calcification induced by VDN, systolic blood pressure was higher than that of the control group. Areas among the elastic fibers of the medial layer in the calcified aorta stained strongly with von Kossa stain for calcium mineral deposits (Figure 1). The calcium content and 45Ca2+ accumulation in calcified aortas were significantly increased relative to controls. Aortic ALP activity and OPN mRNA levels were significantly increased compared with controls (Figure 3). These changes in vascular calcification were in accordance with those noted in previous reports[26]. Interestingly, it was found that in rats with vascular calcification, plasma and aortic H2S content was decreased, and aortic CSE activity and CSE mRNA levels were decreased (Figure 2). These results suggest that the CSE/H2S pathway in calcified vessels was significantly inhibited. NaHS was used as a source of H2S for the following reasons. NaHS dissociates to Na+ and HS in solution, then HS associates with H+ and produces H2S. It does not matter whether the H2S solution is prepared by bubbling H2S gas or by dissolving NaHS. In physiological saline, approximately one-third of the H2S exists as the undissociated form (H2S), and the remaining two-thirds exist as HS at equilibrium with H2S[31]. The use of NaHS enables us to define the concentrations of H2S in solution more accurately and reproducibly than bubbling H2S gas. The influence of <1 mmol/L sodium ions on the electrophysiological experiments was negligible, because saline solution contains 130 mmol/L sodium ions. NaHS at the concentrations used in the present study does not alter the pH. For these reasons, NaHS has been widely used for studies of H2S[32–34]. Administration of NaHS obviously reduced blood pressure in rats with vascular calcification, compared with rats with vascular calcification that did not receive NaHS. Treatment with low and high doses of NaHS decreased systolic blood pressure. NaHS significantly decreased the aortic calcium mineral deposits (Figure 1). The rats treated with low doses of NaHS had reduced vascular calcium content, 45Ca2+ accumulation and ALP activity, and a high dose of NaHS produced better effects than the low dose with respect to these indices. The amount of aortic OPN mRNA were decreased. No significant difference was found in these parameter between the 2 NaHS-treated groups (Table 1, Figure 3) . In the present study, we used NaHS as a donor of H2S, because it is more stable than H2S gas, and does not change the pH value of the plasma[32–34]. Administration of NaHS (2.8 or 14 μmol/kg per d) obviously reduced the elevated blood pressure, calcium overload, and ALP activity in the calcified vessels. von Kossa staining showed that calcium deposition was lessened with NaHS treatment. These results suggest that H2S could significantly inhibit the pathogenesis of vascular calcification.
The biological effect and signal transduction pathway of H2S have not yet been elucidated. Unlike NO and CO, the actions of which are mediated by a second messenger, cGMP, as a physiological regulator of cardiovascular function, H2S is mediated mainly by KATP channel opening[20,35]. The anti-proliferation effect of H2S on vascular smooth muscle cells could be via inhibition of the mitogen-activated protein kinase (MAPK) pathway[20,36]. Mody et al reported that oxygen free radicals, such as H2O2 and oxidized low density lipoprotein were key inducers of vascular calcification[30]. It has been reported that H2S increases the intracellular NADPH:NADP ratio; downregulates some electron trans-porters, ATP-generating genes (including mitochondrial cytochrome oxidase subunits I, II, III, mitochondrial cytochrome C oxidase subunit IV, and ATP synthase subunit d), redox homeostasis genes (including glutathionine S-transferase subunit 8, glutathionine S-transferase M5, metallothionein-2, and metallothionein-1); and decreases the redox environment in IEC-18 cells[37]. H2S might modulate oxygen free radical release and reduce the accumulation of lipid peroxidation products. H2S also directly clears H2O2 and superoxide anions, antagonizes peroxynitrite-mediated damage, and it is considered to be an endogenous antagonist of peroxynitrite[38]. Therefore, H2S could inhibit the development of vascular calcification by regulating oxidative stress.
In the present experiment the endogenous CSE/H2S pathway was obviously downregulated in calcified vessels. CSE is a key enzyme of endogenous H2S generation in vivo[39]. But the transcriptional regulatory mechanism of CSE is still unclear. Wang et al reported that an NO donor also enhances the expression level of CSE and increases H2S production in cultured vascular smooth muscle cells[29]. Our previous work showed that NO production was decreased in calcified vessels[18]. Whether the downregulation of CSE gene expression induced by decreased NO production in calcified vessels is responsible for the diminished H2S generation or not needs further investigation.
The gaseous transmitters, NO, CO and H2S, have some similar cardiovascular effects, and synergistically regulate cardiovascular homeostasis. Our previous work showed that NO and CO are involved in the development of vascular calcification[18,19]. In the present work, we found that H2S also participated in the pathogenesis of vascular calcification. It has been shown that the endogenous production of H2S from rat aortic tissues is enhanced by NO donor treatment[20,29]. The NO donor also enhances the expression level of CSE in cultured vascular SMC. Hosoki et al observed that the vasorelaxant effect of sodium nitroprusside, an NO donor, was enhanced by incubating rat aortic tissues with NaHS[40]. These results suggest that NO, CO and H2S might interact with each other. Elucidation of the functions of and interactions between NO, CO and H2S has important physiological and pathophysiological significance for understanding the pathogenic mechanisms of vascular calcification, and could provide a new target for the prevention and treatment of vascular calcification and related diseases.
References
- Farzaneh FA, Proudfoot D, Shanahan C, Weissberg PL. Vascular and valvar calcification: recent advances. Heart 2001;85:13-7.
- Wallin R, Wajih N, Greenwood GT, Sane DC. Arterial calcification: a review of mechanisms, animal models, and the prospects for therapy. Med Res Rev 2001;21:274-301.
- Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol 2004;15:2959-64.
- Yang H, Curinga G, Giachelli CM. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int 2004;66:2293-9.
- Hofbauer LC, Schoppet M. Osteoprotegerin: a link between osteoporosis and arterial calcification? Lancet 2001;358:257-9.
- Nandalur KR, Baskurt E, Hagspiel KD, Phillips CD, Kramer CM. Calcified carotid atherosclerotic plaque is associated less with ischemic symptoms than is noncalcified plaque on MDCT. Am J Roentgenol 2005;184:295-8.
- Schurgers LJ, Teunissen KJ, Knapen MH, Geusens P, van der Heijde D, Kwaijtaal M, et al. Characteristics and performance of an immunosorbent assay for human matrix Gla-protein. Clin Chim Acta 2005;351:131-8.
- Basalyga DM, Simionescu DT, Xiong W, Baxter BT, Starcher BC, Vyavahare NR. Elastin degradation and calcification in an abdominal aorta injury model: role of matrix metalloproteinases. Circulation 2004;110:3480-7.
- Iba K, Takada J, Yamashita T. The serum level of bone-specific alkaline phosphatase activity is associated with aortic calcification in osteoporosis patients. J Bone Miner Metab 2004;22:594-6.
- Cola C, Almeida M, Li D, Romeo F, Mehta JL. Regulatory role of endothelium in the expression of genes affecting arterial calcifica-tion. Biochem Biophys Res Commun 2004;320:424-7.
- Abedin M, Tintut Y, Demer LL. Vascular calcification. Mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 2004;24:1161-70.
- Zhang B, Tang C, Jiang Z, Qi Y, Pang Y, Du J. Effects of adreno-medullin on vascular calcification in rats. Z Kardiol 2002;91:568-74.
- Qi YF, Wang SH, Zhang BH, Bu DF, Shu TC, Du JB. Changes in amount of ADM mRNA and RAMP2 mRNA in calcified vascular smooth muscle cells. Peptides 2003;24:287-94.
- Wu SY, Zhang BH, Pan CS, Jiang HF, Pang YZ, Tang CS, et al. Endothelin-1 is a potent regulator in vivo in vascular calcification and in vitro in calcification of vascular smooth muscle cells. Peptides 2003;24:1149-56.
- Huang Z, Li J, Jiang Z, Qi Y, Tang C, Du J. Effects of adreno-medullin, C-type natriuretic peptide, and parathyroid hormone-related peptide on calcification in cultured rat vascular smooth muscle cells. J Cardiovasc Pharmacol 2003;42:89-97.
- Jeziorska M. Transforming growth factor-betas and CD105 expression in calcification and bone formation in human atherosclerotic lesions. Z Kardiol 2001;90:23-6.
- Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 2000;102:2636-42.
- Zhang BH, Wu SY, Pang YZ, Li JX, Tang CS, Du JB. Effects of L-arginine and nitric oxide inhibitor on vascular calcification in rats. Basic Clin Med 2003;23:599-603.
- Zhang B, Wang S, Pang Y, Tang C, Du J. Alteration of heme-oxygenase-carbon monoxide pathway in calcified rat vascular smooth muscle cells. Z Kardiol 2004;93:109-15.
- Geng B, Du JB, Tang CS. Endogenous H2S-a new gas transmitter. Prog Physiol Sci 2002;33:255-8.
- Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J, et al. H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun 2004;313:362-8.
- Du J, Hui Y, Cheung Y, Bin G, Jiang H, Chen X, et al. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart Vessels 2004;19:75-80.
- Zhong G, Chen F, Cheng Y, Tang C, Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens 2003;21:1879-85.
- Qingyou Z, Junbao D, Weijin Z, Hui Y, Chaoshu T, Chunyu Z. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hyper-tension. Biochem Biophys Res Commun 2004;317:30-7.
- Chen XB, Du JB, Geng B, Jiang HF, Tang CS. Changes in arterial hydrogen sulfide (H2S) content during septic shock and endotoxin shock in rats. Basic Clin Med 2003;23:384-7.
- Niederhoffer N, Bobryshev YV, Lartaud-Idjouadiene I, Giummelly P, Atkinson J. Aortic calcification produced by vitamin D3 plus nicotine. J Vasc Res 1997;34:386-98.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248-54.
- Qi YF, Shi YR, Bu DF, Pang YZ, Tang CS. Changes of adreno-medullin and receptor activity modifying protein 2 (RAMP2) in myocardium and aorta in rats with isoproterenol-induced myocardial ischemia. Peptides 2003;24:463-8.
- Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J 2002;16:1792-8.
- Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radical Biol Med 2001;31:509-19.
- Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol 1992;32:109-34.
- Beauchamp RO Jr, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol 1984;13:25-97.
- Warenycia MW, Steele JA, Karpinski E, Reiffenstein RJ. Hydrogen sulfide in combination with taurine or cysteic acid reversibly abolishes sodium currents in neuroblastoma cells. Neurotoxi-cology 1989;10:191-9.
- Kombian SB, Reiffenstein RJ, Colmers WF. The actions of hydrogen sulfide on dorsal raphe serotonergic neurons in vitro. J Neurophysiol 1993;70:81-96.
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 2001;20:6008-16.
- Yang G, Sun X, Wang R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J 2004;18:1782-4.
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J 2004;18:1165-7.
- Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem 2004;90:765-8.
- Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, et al. Murine cystathionine gammalyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem 2004;381:113-23.
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 1997;237:527-31.