
Recombinant approaches to IgG-like bispecific antibodies
Introduction
Antibodies quickly evolve by recombination, somatic mutation, and clonal selection in vivo to acquire the exquisite specificity and high affinity needed for an effective immune response. The modular structure of antibodies that permits in vivo reorganization also makes them exceptionally amenable to molecular manipulation, and facilitates the development of desirable properties. As a result, the field of antibody engineering has developed rapidly in recent years, as researchers exploit the ease with which antibodies can be genetically modified to develop ever more powerful therapeutics. So far, 18 antibody-based products have been approved by the FDA for therapeutic applications, including 8 for oncology indications. While the inoculation of animals with antigen and high-throughput screening of hybridoma clones is still common practice, in vitro selection is rapidly becoming the norm. Through the use of synthetic or semi-synthetic libraries in microscopic selection techniques (such as phage display[1], yeast surface display[2], and ribosomal display[3]), identifying an antibody to a given target has become a routine, although by no means trivial, matter.
Once an antibody specific to a particular antigen is identified, numerous mechanisms of action can be exploited for therapy. These mechanisms include: (1) as an antigen (growth factor or cytokine) sequestrant, the antibody binds a soluble antigen and prevents it from interacting with other molecules, such as its receptors. Avastin (Genentech, South San Francisco, CA), an anti-angiogenic therapy for colon cancer, binds vascular endothelial growth factor (VEGF) and blocks its interaction with the receptors[4]. Remicade (Centocor, Malvern, PA), a treatment for auto-immune disorders such as Crohn’s disease and rheumatoid arthritis, works by sequestering tumor necrosis factor-α[5]. A number of antibodies to sequester anthrax toxin[6] and botulinum toxin[7] are also being developed; (2) as a receptor antagonist, the antibody binds a cell surface receptor and inactivates it by blocking the binding site of an activating ligand. Erbitux (ImClone, New York, NY), an antibody for the treatment of colon cancer, binds epidermal growth factor receptor (EGFR) at the EGF-binding site and blocks activation by both EGF and transforming growth factor-α[8]. Alternatively, the antibody may not directly block ligand/receptor interaction but rather exerts its effects by preventing receptor dimerization/multimerization, which is required for activation. For example, Omnitarg, also known as 2C4 (Genentech), an anti-HER2 antibody currently under clinical development, is believed to inhibit tumor cell growth by blocking HER2 from dimerizing with EGFR and HER3 (there is no ligand identified so far for HER2)[9]; (3) as an agonist, the antibody binds to and cross-links multiple membrane bound receptors, mimicking the function of a natural ligand, and activating the receptor. There are a number of antibodies that mimic the function of Apo2L (ligand) and trigger apoptosis by activation of death receptor 5[10,11]; (4) as an effector function activator, the antibody binds a cell surface target and acts as an immune system identifier for antibody dependent cellular cytotoxicity (ADCC) or complement-mediated cytotoxicity (CMC). Rituxan (Genentech), an antibody for the treatment of CD20-positive, B-cell non-Hodgkin’s lymphoma[12], and Herceptin (Genentech), an antibody for the treatment of HER2 positive metastatic breast cancer[13,14], are postulated to exert their effect in part by this mechanism[15]; (5) as a chemotherapy or radiotherapy adjunct, the antibody acts as a carrier molecule to deliver an attached chemotherapeutic agent or toxin, or radioisotope to cells displaying a specific antigen. A number of antibody conjugates have been approved by the FDA for oncology indications, including Mylotarg (Wyeth, Madison, NJ), an anti-CD33 antibody-calicheamicin conjugate for the treatment of CD33 positive acute myeloid leukemia; Zevalin (Biogen Idec, Cambridge, MA), a 90Y-labeled anti-CD20 antibody, and Bexxar (GlaxoSmithKline, Brentford, UK), an 131I-labeled anti-CD20 antibody for non-Hodgkin’s lymphoma[16]; (6) as a means to redirect cytotoxic agents or immune effector cells to target sites, such as tumors, in the form of a bispecific antibody (BsAb), which is discussed in detail in the present paper.
Bispecific antibodies
BsAb are antibody-based molecules that can simultaneously bind two separate and distinct antigens (or different epitopes of the same antigen). The primary use of BsAb has been to redirect cytotoxic immune effector cells for enhanced killing of tumor cells by ADCC. In this context, one arm of the BsAb binds an antigen on the tumor cell, and the other binds a determinant expressed on effector cells, such as CD3, CD16, or CD64, which are expressed on T lympho-cytes, natural killer (NK) cells, or other mononuclear cells[17–20]. By cross-linking tumor and effector cells, the BsAb not only brings the effector cells within the proximity of the tumor cells but also simultaneously triggers their activation, leading to effective tumor cell-killing. Preliminary but promising clinical benefits have been observed in a number of early stage trials[21]. In addition, BsAb has also been used to enrich the tumor/normal tissue localization ratio of chemo- or radiotherapeutic agents. In this setting, one arm of the BsAb binds an antigen expressed on the cell targeted for destruction, and the other arm binds a chemotherapeutic drug, radioisotope, or toxin. The naked BsAb is administered first, and after sufficient time has passed for the BsAb to bind tumor cells and to clear from normal tissue, the cytotoxic molecule is delivered, with rapid accumulation in the tumor, because of its affinity for the tumor bound BsAb[21–25]. Recently, a novel concept has emerged - the development of BsAb that target two tumor-associated antigens (eg, growth factor receptors) for down-regulation of multiple distinct cell proliferation/survival pathways, which provides enhanced antitumor activity[26–28].
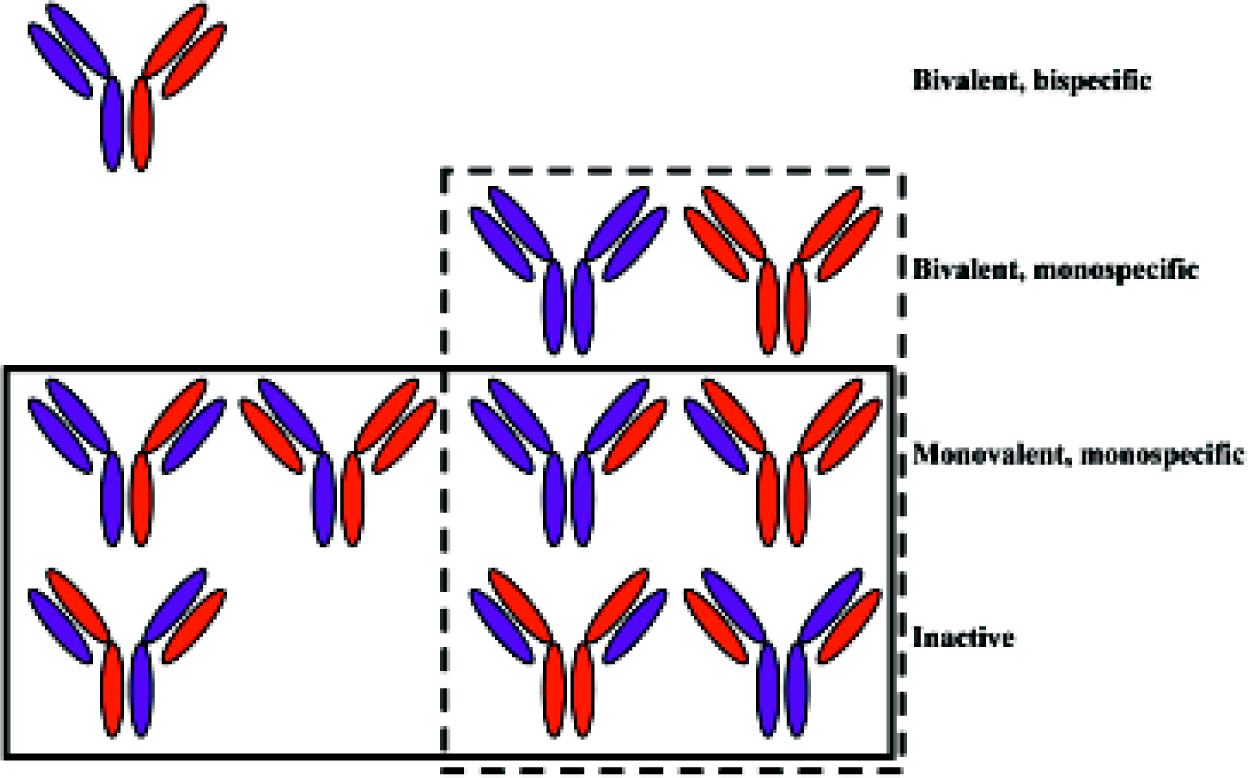
A major obstacle in the general development of BsAb has been the difficulty of producing materials of sufficient quality and quantity for both preclinical and clinical studies. Initially, the main route to the production of BsAb was by co-expression of both the light chains (LC) and both the heavy chains (HC) of two parent antibodies of different specificities (antibody A and antibody B) in a single cell through either the hybrid hybridoma technique[29] or DNA co-transfection. Unfortunately, assuming that all the four polypeptide chains are equally expressed and there is no pairing preference between any particular LC and HC, in addition to the desired heterodimeric BsAb product (LCA-HCA plus LCB-HCB), there is also a large number of undesired products formed from the ten molecules that result from the 16 permutations LC and HC pairings (Figure 1). Conse-quently, the desired binding-competent BsAb are a minor product (in theory, an eighth of the total), and purification from the other products is very difficult. Another traditional method for BsAb production is chemical conjugation of two antibodies (or their fragments) of different specificities[30], although this method is by no means simple. Furthermore, the chemical modification process may inactivate the antibody or promote aggregation. As purification from undesired products remains difficult, the resulting low yield and poor quality BsAb makes this process, like the hybrid hybridoma and DNA co-transfection, unsuitable for the large scale production required for clinical development.
Thus the major requirements of efficient BsAb production are: (1) a novel structural format that promotes or obligates the formation of homogenous, bispecific proteins; and (2) an efficient expression system in prokaryotic or eukaryotic cells that leads to high-level production. Significant progress has been made in the past decade towards the development of BsAb fragments[31,32], which have various advantages and disadvantages. Firstly, they are smaller than full length IgG, so they have better solid tumor penetration rates, but their small size and lack of an intact Fc also results in rapid clearing from circulation, leading to a short in vivo half-life. Secondly, the BsAb fragments do not require glyco-sylation, so they can be produced in high yield in bacteria. Compared to the full length IgG-like BsAb, these fragments are, however, incapable of promoting effector function such as ADCC and CMC (unless one arm of the BsAb fragments is specifically targeted to bind an effector cell determinant or certain components of the complement proteins, eg, C1q). The engineering and application of various BsAb fragments have been discussed in a recent review by Kontermann[33]. Our review will thus focus on the latest advancements and ongoing developments in recombinant production of full-length, IgG-like BsAb.
Recombinant approaches to IgG or IgG-like BsAb
Increasing heterodimer: homodimer ratio
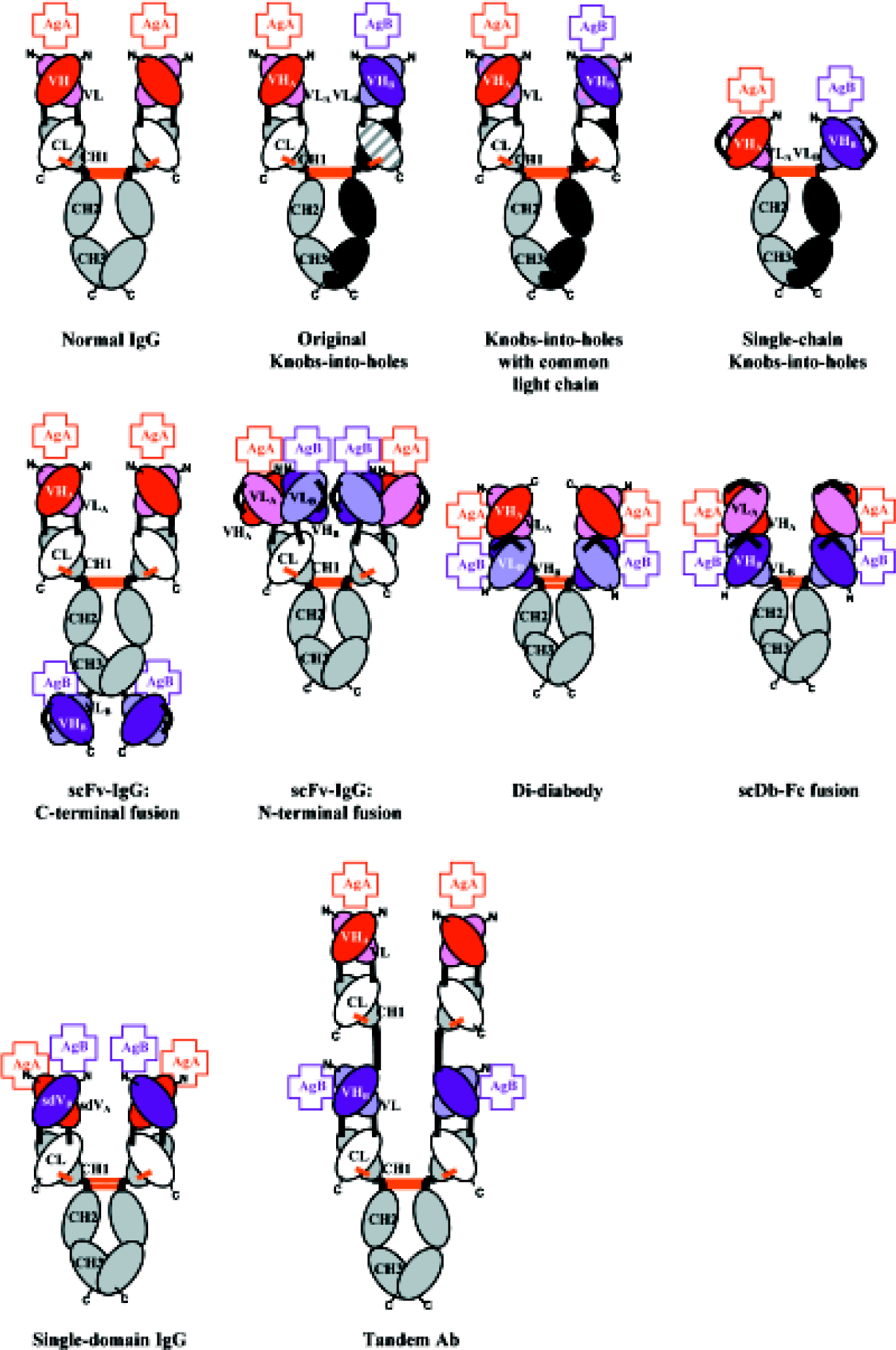
“Knobs-into-holes” BsAb IgG Because one of the major barriers to the production of BsAb in a single cell system, such as hybrid hybridoma, is the undesired formation of HC homodimers (Figure 1), a first and obvious solution is to re-engineer the CH3 domain of the Fc so as to favor HC heterodimerization over homodimerization. This concept, first developed by Carter et al at Genentech[34], is based on the idea that simple mutations can be introduced into one half of the CH3 dimer, such that the steric complementarity required for CH3/CH3 association obligates the mutated CH3 domain to pair with a CH3 domain that has different, accommodating mutations (Figure 2). Specifically, a “knob” mutation (T366W in the CH3 domain, chosen by inspection of the crystal structure of the Fc[35]) is made into one HC to introduce a larger residue at the CH3 dimer interface and create a steric barrier to homodimerization. To promote heterodi-merization, an accommodating “hole” (Y407A) mutation is engineered into the CH3 domain of the other HC. Co-expression of these two HC results in the formation of a mostly (92%) heterodimeric product, but with a stability that is significantly lower than that of the wild-type (knob: T366W+ hole: Y407A mutant, Tm=65.4°C; wild-type, Tm=80.4°C). To optimize and stabilize the heterodimerized molecules, variants with enhanced stability were selected from a phage display library in which residues near the “hole” (T366, L368, Y407) were randomized[36]. The resulting variant (knob: T366W; hole: T366S, L368A, Y407V) has an increased Tm (69.4°C) and formed predominantly heterodimers, when co-transfected in 293 cells. This example clearly demonstrates the utility of the knob-into-holes approach. It is possible there are other sets of residues within the CH3 domains that could be mutated/optimized to further increase BsAb stability and the heterodimer: homodimer ratio.
While this approach solves the HC homodimerization problem, and reduces the total number of potential LC/HC pairings in a co-transfected single host from ten to four (Figure 1) thus, in theory, increasing the production of the functional BsAb from 12.5% to 25%, it does not address the equally problematic mispairing of LC and HC from two different antibodies (eg, LCA-HCB and LCB-HCA mispairing, instead of the correct LCA-HCA and LCB-HCB pairing). This can be resolved by similarly redesigning the LC-HC interaction to incorporate knobs and holes at the VL-VH interface[37]. Based on inspection of the VL-VH interface of the anti-HER2 Fab fragment, 4D5 Fab[38], knob mutations and the complementary hole mutations were designed for a diabody fragment containing the Fvs of 4D5 and an anti-CD3 antibody. For one of the variants (termed “v5”; knob: VH-V37F, L45W; hole: VL-Y87A, F98M) 92% of the products were hetero-dimers, and near wild-type affinity for antigens was retained. Although these mutations were used to promote hetero-dimerization in a bispecific diabody, the principle should translate easily to a full length IgG format.
Common light chain BsAb IgG An alternative approach to solve the LC-HC mispairing problem is to construct BsAb using two antibodies of different specificities that share an identical LC[39] (Figures 1, 2). In a “proof-of-concept” study, a large scFv phage display library was used to screen for antibodies to a variety of antigens. The VL fragments of selected scFv were compared and those with identical amino acid sequences, but paired with different VH fragments for different antigen binding specificity, were selected to create BsAb. Functional IgG-like BsAb were formed with high yield (>95% of total IgG products) when a single LC was co-transfected in a host with two different HC that were engineered to incorporate the original knobs and holes mutations (knob: T366W, hole: T366S, L368A, Y407V) and a heterodimer promoting/stabilizing disulfide bond (S354C, Y349’C).
This highly engineered system demonstrates that the knobs-into-holes principle, when combined with a common light chain, can be used to effectively produce a near homogenous population of BsAb IgG. It also suggests that there are multiple ways in which the knob-into-holes approach can be applied, and that BsAb with even higher ratios of heterodimer formation may be achieved with further engineering. However, an obvious drawback of this method is that the inclusion of multiple mutations in the CH3 domains might pose an immunogenic risk in a therapeutic setting. Furthermore, it requires the identification of antibodies with common LC, which is rare, particularly for high-affinity antibodies.
Single chain Fv-Fc “knobs-into-holes” BsAb The LC-HC mispairing problem may also be circumvented by fusing the VL and VH in a single-chain Fv (scFv) format[40]. In this construct, an anti-HER2 scFv is fused to an Fc with the “knob” mutation T366Y, and an anti-CD16 scFv is fused to an Fc with the “hole” mutation Y407T (Figure 2). Efficient pairing of the two chains, through the knobs-into-holes mechanism, was demonstrated (but not quantitated), and specificity for both antigens was confirmed by cell surface binding analysis. The scFv-Fc knobs-into-holes molecule is also able to crosslink the two target antigens, as shown by its ability to induce higher NK cell-mediated cytotoxicity than a monospecific anti-HER2 antibody alone. One could hypothesize that this format, which is slightly smaller (120 kDa) than a normal IgG (150 kDa), might provide slight gains in tumor penetration, while maintaining the longer half-life and effector functions provided by the intact Fc region.
IgG-scFv fusions To circumvent the LC-HC mispairing and HC homodimerization issues completely, a number of formats have been developed to construct and produce BsAb that achieve their bi-specificity and product homogeneity not by molecular alteration of the component domains, but by direct addition of a new antigen binding specificity to a fully functional antibody or an antibody-like molecule,such as an IgG antibody or a scFv-Fc fusion protein (Figure 2).
C-terminal scFv fusion One approach is to fuse a scFv specific for one antigen to the carboxy terminus of a full-length IgG antibody specific for another antigen, creating a tetravalent bispecific IgG-like antibody[41] (Figure 2). The specificity at the N-terminal end (in this case, for dextran) is defined by a normal Fab. The specificity at the C-terminal end (in this case, for dansyl) is conferred by an anti-dansyl scFv. The fusion construct has an affinity approximately 10-fold lower for dansyl than expected (as compared to its parent IgG), primarily because of a slower on-rate. This may be a result of steric hindrance from the rest of the IgG, an inability of both scFv fragments to bind antigen simultaneously (whether all four binding sites were occupied was not tested), or an artifact resulting from conversion of the original anti-dansyl IgG to scFv format. Regardless, the constructs in both a full-length IgG-like format (VLA-CL plus VHA-CH1-CH2-CH3-scFvB)2 and a F(ab’)2-like format (VLA-CL plus VHA-CH1-scFvB)2 were capable of binding both target antigens. Furthermore, the IgG-like version is capable of binding C1q, presumably through the intact Fc fragment, although it is unable to trigger a complete complement cascade. This format has the potential to allow the creation of tetravalent molecules with some effector function, but the full potential of the latter remains to be investigated.
N-terminal scFv fusion In an alternative approach, the two scFv of different specificities are fused, respectively, to the N-termini of the constant light (CL) and the first-constant heavy (CH1) domains of an IgG: (scFvA)-CL and (scFvB)-CH1-CH2-CH3. Co-expression of the two polypeptide chains in a single host results in the formation of a tetravalent bispecific molecule, Bs(scFv)4-IgG (Figure 2)[42]. Only homogenous BsAb are generated as a result of the natural heterodimerization between the CL and the CH1 domains. Similar to the C-terminal fusion, this molecule is also amenable to truncation: a bivalent bispecific Fab-like molecule can be created without the Fc fragment. The ability of this construct to crosslink antigen (in this case, two distinct epitopes on vascular endothelial growth factor receptor 2 (VEGFR2) was demonstrated, but it is not clear whether all four binding sites are or can be occupied simultaneously.
Two other Bs(scFv)4-IgG molecules were recently constructed, using the same two scFv fragments directed against EGFR and insulin-like growth factor receptor (IGFR), but in different fusion orientations[28]. Both constructs, scFvanti-EGFR-CL plus scFvanti-IGFR-CH1-CH2-CH3, or scFvanti-IGFR-CL plus scFvanti-EGFR-CH1-CH2-CH3, blocked EGF and IGF from binding their respective receptors and inhibited signal transduction pathways activated by both EGF and IGF, whereas a monospecific antibody only inhibited the pathway stimulated by a single ligand. In addition, the BsAbs inhibited tumor cell proliferation in vitro at a level that is on par with the combination of the two parental IgG antibodies. Furthermore, both Bs(scFv)4-IgG demonstrated very good stability when incubated in vitro in mouse serum at 37 ºC for 7 d. We believe that this Bs(scFv)4-IgG format should be applicable to BsAb construction from two antibodies directed against any pair of antigens. A drawback of the format is its low expression level in mammalian cells, probably because of both its large size (~ 200kDa) and structural complexity.
Diabody-Fc fusions Another method by which homogenous populations of IgG-like BsAb can be constructed is by replacement of the Fab fragment with a bispecific diabody. Diabodies are a derivative of the scFv construct[43]. An scFv is composed of a VH and a VL domain connected by a flexible linker of approximately 15 amino acids, such as (Gly4Ser)3, that permits self assembly into an antigen binding competent form. If the linker is shortened to 5 amino acids, such as Gly4Ser, self assembly is impossible, and two scFv interact with each other to form a bivalent molecule of two interlinked polypeptides, the VL of one chain associating with the VH of the other. If VL and VH with specificities for different antigens comprise the diabody; namely, VHA-VLB and VHB-VLA (the two so-called “cross-over” scFv), bispecific bivalent diabodies are formed, with one binding site for each antigen. In addition to assembling as functional hetero-dimers, the cross-over scFv can also assemble as non-functional homodimers. Fortunately, purification of properly heterodimerized molecules can easily be achieved by one round of affinity chromatography. And as noted earlier, the knobs-into-holes technique can be used to re-engineer the Fv interface to promote the correct heterodimeric VH-VL pairing[37]. Diabodies have shown to be useful for antigen cross linking[43,44], and their small size is valuable for tumor penetration[45]. However, like many other smaller bispecific fragments, diabodies lack functional Fc domains and the corresponding effector function[46]. Recently, they have been fused to the Fc domain of an IgG to create tetravalent IgG-like BsAb.
Single chain diabody-Fc fusion Kipriyanov et al have developed a “single chain” diabody (scDb) by fusing both “cross-over” scFv of a bispecific diabody with a flexible linker[47]. This construct is fused to an Fc fragment (or just a CH3 domain) to create a tetravalent bispecific IgG-like molecule (Figure 2). In this format, one polypeptide with six domains is produced: VHA-VLB-VHB-VLA-CH2-CH3, which then assembles into IgG-like dimers through the Fc domains[48]. The scDb-Fc is bispecific and bivalent for both antigens, and has a full Fc (although effector function activity was not tested). The stability of the scDb-Fc fusion was not reported, but the multiple exposed non-human polypeptide linkers within the molecule may not only subject the BsAb to proteolytic cleavage, but may also introduce a potential immunogenic risk, thus lowering the utility of the molecule in vivo. Finally, this format lacks sufficient levels of expression (~5 mg/L) to make a practical transition to a therapeutic molecule.
Di-diabody An alternative diabody-Fc fusion format is the so-called “di-diabody”. In this construct, one half of a diabody; namely, one “cross-over” scFv, is fused to the Fc domain, creating a “heavier chain” (VLA-VHB-CH2-CH3) and the other “cross-over” scFv associates with it as a “lighter chain” (VLB-VHA)[27]. The heavier and lighter chains assemble with each other through the VH-VL interfaces, and two heavier chains homodimerize through the Fc regions to form an IgG-like tetravalent BsAb (Figure 2). Although lighter chains can homodimerize to form non-functional diabodies, they are easily removed when the full length functional di-diabody is purified by protein A chromatography. A similar but smaller di-diabody construct can also be created by just using the CH3 domain for dimerization, namely, VLB-VHA plus VLA-VHB-CH3[49]. As a precautionary measure to avoid potential immunogenicity in human therapy, a human sequence, the first 5 amino acids of the human IgG CL(Kappa) domain, is used as the linker between the variable domains[44], instead of a “standard” Gly4Ser linker. A di-diabody that binds both EGFR and IGFR was constructed using the variable domains of an anti-EGFR and an anti-IGFR antibody. The di-diabody has affinity for its antigens, EGFR and IGFR, similar to that of the parental antibodies from which the VL and VH were derived. The di-diabody blocks both EGF and IGF from binding their respective receptors and down-regulates the signal transduction pathways activated by each ligand. In addition, the di-diabody induces efficient ADCC activity against tumor cells that express EGFR and/or IGFR, indicating that the di-diabody possesses an intact and unhindered Fc domain. It also has an in vivo half-life that is equivalent to that of an intact human IgG. Finally, the di-diabody effectively inhibits the growth of two different human tumor xenografts in nude mice. Unlike the Bs(scFv)4-IgG format discussed previously, the di-diabody construct is expressed in mammalian cells at much higher levels (>400 mg/L in unoptimized conditions), which could greatly facilitate the transition from “proof-of-concept” to therapeutic application. Unfortunately, the di-diabody construct has a tendency to form inactive molecules in vivo that lack the lighter chain, a result of dissociation between the heavier and the lighter chains (caused by the inherent instability of diabodies) followed by the rapid clearance of the lighter chain from the circulation. Hopefully, this shortcoming will be surmounted by either the introduction of disulfide bonds[37,50,51] or improved packing[52] to stabilize the VL-VH interfaces in the diabody.
Future perspective
Other novel IgG-like BsAb constructs
BsAb IgG using single domain antibodies as building blocks The aforementioned methods use some form of Fv, comprised of a VL and a VH, to bind antigen. An emerging and promising novel approach in which BsAb can be constructed is to use the VL and VH as independent binding units. It has been observed that some camel antibodies are composed of only heavy chains[53]. This observation has led to the development of human “single domain” antibody fragments, in which a VL or a VH alone comprises the binding unit[54,55]. These fragments can then be used to construct tetravalent IgG-like BsAb by fusing a single domain of one specificity to CL and a single domain of a different specificity to the CH1 of an IgG. Preferably, one single domain would be derived from a VL, and the other from a VH, to provide increased stability using a VL-VH interface, in addition to that of the CL-CH1 interface (ie, VLA-CL plus VHB-CH1-CH2-CH3) (Figure 2), but an IgG-like molecule with bispecific binding capacity derived from any single domain combination could be imagined (ie, VLA-CL plus VLB-CH1-CH2-CH3, or VHA-CL plus VHB-CH1-CH2-CH3).
Tandemabs An interesting application of antibody engineering that could readily be applied to the construction of BsAb is an expansion of the multivalent Fab constructs developed by Presta et al at Genentech[56]. Based on the observation that cross-linking increases the biological efficacy of some antibodies, a series of multivalent antibody constructs were developed from the interaction of an identical LC with tandem repeats of the VH-CH1 unit fused to the Fc. If one were to construct a pseudo-tandem repeat of the form VHA-CH1-VHB-CH1-CH2-CH3, and pair that with a common LC unit VL-CL, one could achieve tetravalent (or possibly higher order) bispecific (or tri-specific) antibodies (Figure 2). Again, the challenge here is to identify antibodies of different specificities that share identical LC.
Novel applications of BsAb As noted earlier, most of the efforts in BsAb development have been focused on the use of BsAb as means to redirect either effector cells or cytotoxic agents, including chemotherapeutic drugs, radioiso-topes, and toxins, to the sites of destruction, such as tumors. There are a number of novel benefits of BsAb that can be exploited for more efficacious human therapy.
Binding avidity enhancement One very valuable benefit of BsAb is the enhanced avidity they pose for their antigen[57]. In addition to having intrinsic high affinity on a binding unit (ie, a Fab) to antigen basis, normal IgG antibodies also exploit the avidity effect to increase their association with antigens as a result of their bivalent binding towards the targets. Except for the knob-into-holes BsAb format, which is monovalent for each antigen, all the IgG-like BsAb discussed here are bivalent for each antigen. Consequently, these IgG-like BsAb, once bound to cell-surface antigens, are expected to dissociate at a very slow rate: two dissociation events must occur simultaneously for the BsAb to be free from the cell. High binding affinity (or avidity) is usually beneficial and, in some cases, may even be required for the biological activity of a therapeutic antibody. In addition, bivalent binding to a target, particularly a cell-surface receptor, is under many circumstances a prerequisite for antibody function, such as cross-linking the receptors in order to stimulate activation, to induce apoptosis, or to promote receptor internalization.
Epitope cross-linking for acquired antagonistic activity A BsAb directed against two separate epitopes on the same antigen molecule may not only provide the benefit of enhanced binding avidity (because of bivalent binding), but may also acquire novel properties that are not associated with either of the parent antibodies. For example, we have demonstrated that a bispecific diabody that binds two distinct epitopes on VEGFR2 cross-links the epitopes and effectively blocks the binding of VEGF to the receptor, whereas the parent scFv from which the bispecific diabody is derived do not, on their own or in combination, block VEGF binding[57]. Cross-linking two separate epitopes within the same receptor molecule may introduce new steric hindrance for ligand binding, and/or induce conformation changes in the receptor, preventing it from binding ligand. It will be interesting to see if this principle is applicable to antibodies to other receptors for which ligand-blocking antibodies are difficult to identify.
Fine-tuning antibody specificity towards tumor cells An emerging and intriguing concept is the use of BsAb to further fine-tune the specificity of antitumor antibodies to target cells. In this context, a BsAb is constructed from two antibodies directed against different tumor antigens, each of low to moderate binding affinity. Only simultaneous binding (or cross-linking) to the two target antigens on the same tumor cell surface by BsAb leads to strong association (ie, high avidity resulting from bivalency), which is required to trigger biological processes, such as ADCC, CMC, and down-regulation of signaling pathways. In contrast, weak association, which may not be sufficient to induce any meaningful cellular activity, would result from monovalent binding of the BsAb to cells that only express one of the target antigens. This is significant as most of the targets currently being used as tumor cell identifiers are, in fact, not truly “tumor-specific” but rather “tumor-associated”, that is, they are also expressed in certain normal tissues/cells, albeit at lower density than in tumor cells. By identifying and constructing BsAb to pairs of targets simultaneously expressed on a given type of tumor, one could expect enhanced antibody specificity towards the targeted cells while sparing normal cells of unwanted side effects.
Simultaneous blockade of two signaling pathways Perhaps the most promising application of BsAb is the ability to simultaneously block the signaling pathways of two targets with one molecule[26–28,58]. As demonstrated with the anti-EGFR×anti-IGFR BsAb, either in the form of di-diabody or Bs(scFv)4-IgG[27,28], it is possible to achieve the effects of administering two antibodies with one IgG-like BsAb. This provides a promising alternative to the development of antibody combination therapies, as in the latter each antibody has to be approved separately by regulatory agencies before being approved as a combination. Furthermore, if BsAb can be produced in similar quantities as normal IgG, then significant cost savings can be achieved.
Conclusion
Recently, developments in the design of IgG-like BsAb have provided some success in overcoming the major obstacles to BsAb production. It is now possible to use a novel structural format that promotes or obligates the formation of homogenous, bispecific proteins, and some of these constructs can be efficiently produced in eukaryotic cells in quantities sufficient for clinical development. Furthermore, IgG-like BsAb offer the benefits of normal IgG, including long half-life and native effector function, along with the additional capability of being able to act as two drugs in one by simultaneously addressing two disease-relevant targets in a highly specific manner, thus making them in many ways superior to BsAb fragments. We believe that IgG-like BsAb will undoubtedly challenge monospecific antibodies as the champion of therapeutic proteins in coming years.
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; BsAb, bispecific antibody; CL, constant domain of light chain; CH, constant domain of heavy chain; CMC, complement-mediated cytotoxicity; Fab, antigen binding fragment; Fc, crystallized fragment; Fv, variable fragment (VL+VH); EGFR, epidermal growth factor receptor; HC, heavy chain; IGFR, insulin-like growth factor receptor; LC, light chain; scFv, single-chain variable fragment (VL and VH tethered by a 15 amino acid linker); VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; VH, variable heavy domain; VL, variable light domain.
References
- Sidhu SS. Phage display in biotechnology and drug discovery. New York (NY): Marcel Dekker; 2005.
- Colby DW, Kellogg BA, Graff CP, Yeung YA, Swers JS, Wittrup KD. Engineering antibody affinity by yeast surface display. Methods Enzymol 2004;388:348-58.
- Lipovsek D, Pluckthun A. In-vitro protein evolution by ribosome display and mRNA display. J Immunol Methods 2004;290:51-67.
- Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 2004;3:391-400.
- Knight DM, Trinh H, Le J, Siegel S, Shealy D, McDonough M, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol 1993;30:1443-53.
- Maynard JA, Maassen CB, Leppla SH, Brasky K, Patterson JL, Iverson BL, et al. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol 2002;20:597-601.
- Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, et al. Potent neutralization of botulinum neurotoxin by recombinant oligoclonal antibody. Proc Natl Acad Sci USA 2002;99:11346-50.
- Li S, Schmitz K, Jeffery PD, Wiltzius J, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab/Erbitux. Cancer Cell 2005. in press.
- Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004;5:317-28.
- Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med 2001;7:954-60.
- Yagita H, Takeda K, Hayakawa Y, Smyth MJ, Okumura K. TRAIL and its receptors as targets for cancer therapy. Cancer Sci 2004;95:777-83.
- McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998;16:2825-33.
- Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol 1999;17:2639-48.
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783-92.
- Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med 2000;6:443-6.
- Silverman DH, Delpassand ES, Torabi F, Goy A, McLaughlin P, Murray JL. Radiolabeled antibody therapy in non-Hodgkins lymphoma: radiation protection, isotope comparisons and quality of life issues. Cancer Treat Rev 2004;30:165-72.
- Xiong D, Xu Y, Liu H, Peng H, Shao X, Lai Z, et al. Efficient inhibition of human B-cell lymphoma xenografts with an anti-CD20×anti-CD3 bispecific diabody. Cancer Lett 2002;177:29-39.
- Kipriyanov SM, Cochlovius B, Schafer HJ, Moldenhauer G, Bahre A, Le Gall F, et al. Synergistic antitumor effect of bispecific CD19×CD3 and CD19×CD16 diabodies in a preclinical model of non-Hodgkin’s lymphoma. J Immunol 2002;169:137-44.
- James ND, Atherton PJ, Jones J, Howie AJ, Tchekmedyian S, Curnow RT. A phase II study of the bispecific antibody MDX-H210 (anti-HER2×CD64) with GM-CSF in HER2+ advanced prostate cancer. Br J Cancer 2001;85:152-6.
- Baeuerle PA, Kufer P, Lutterbuse R. Bispecific antibodies for polyclonal T-cell engagement. Curr Opin Mol Ther 2003;5:413-9.
- Cao Y, Lam L. Bispecific antibody conjugates in therapeutics. Adv Drug Deliv Rev 2003;55:171-97.
- Corvalan JR, Smith W, Gore VA. Tumour therapy with Vinca alkaloids targeted by a hybrid-hybrid monoclonal antibody recognising both CEA and Vinca alkaloids. Int J Cancer Suppl 1988;2:22-5.
- Ford CH, Osborne PA, Rego BG, Mathew A. Bispecific antibody targeting of doxorubicin to carcinoembryonic antigen-expressing colon cancer cell lines in vitro and in vivo. Int J Cancer 2001;92:851-5.
- Ferrini S, Sforzini S, Canevari S. Bispecific monoclonal antibodies for the targeting of type I ribosome-inactivating proteins against hematological malignancies. Methods Mol Biol 2001;166:177-92.
- Chang CH, Sharkey RM, Rossi EA, Karacay H, McBride W, Hansen HJ, et al. Molecular advances in pretargeting radio-imunotherapy with bispecific antibodies. Mol Cancer Ther 2002;1:553-63.
- Lu D, Jimenez X, Zhang H, Wu Y, Bohlen P, Witte L, et al. Complete inhibition of vascular endothelial growth factor (VEGF) activities with a bifunctional diabody directed against both VEGF kinase receptors, fms-like tyrosine kinase receptor and kinase insert domain-containing receptor. Cancer Res 2001;61:7002-8.
- Lu D, Zhang H, Koo H, Tonra J, Balderes P, Prewett M, et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J Biol Chem 2005. [Epub ahead of print].
- Lu D, Zhang H, Ludwig D, Persaud A, Jimenez X, Burtrum D, et al. Simultaneous blockade of both the epidermal growth factor receptor and the insulin-like growth factor receptor signaling pathways in cancer cells with a fully human recombinant bispecific antibody. J Biol Chem 2004;279:2856-65.
- Milstein C, Cuello AC. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983;305:537-40.
- Brennan M, Davison PF, Paulus H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 1985;229:81-3.
- Carter P, Ridgway J, Zhu Z. Toward the production of bispecific antibody fragments for clinical applications. J Hematother 1995;4:463-70.
- Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR, Hudson PJ. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J Immunol Methods 2001;248:47-66.
- Kontermann RE. Recombinant bispecific antibodies for cancer therapy. Acta Pharmacol Sin 2005;26:1-9.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996;9:617-21.
- Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981;20:2361-70.
- Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol 1997;270:26-35.
- Zhu Z, Presta LG, Zapata G, Carter P. Remodeling domain interfaces to enhance heterodimer formation. Protein Sci 1997;6:781-8.
- Eigenbrot C, Randal M, Presta L, Carter P, Kossiakoff AA. X-ray structures of the antigen-binding domains from three variants of humanized anti-p185HER2 antibody 4D5 and comparison with molecular modeling. J Mol Biol 1993;229:969-95.
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, et al. An efficient route to human bispecific IgG. Nat Biotechnol 1998;16:677-81.
- Xie Z, Guo N, Yu M, Hu M, Shen B. A new format of bispecific antibody: highly efficient heterodimerization, expression and tumor cell lysis. J Immunol Methods 2005;296:95-101.
- Coloma MJ, Trinh KR, Wims LA, Morrison SL. The hinge as a spacer contributes to covalent assembly and is required for function of IgG. J Immunol 1997;158:733-40.
- Zuo Z, Jimenez X, Witte L, Zhu Z. An efficient route to the production of an IgG-like bispecific antibody. Protein Eng 2000;13:361-7.
- Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA 1993;90:6444-8.
- Zhu Z, Zapata G, Shalaby R, Snedecor B, Chen H, Carter P. High level secretion of a humanized bispecific diabody from Escherichia coli. Biotechnology (NY) 1996;14:192-6.
- Colcher D, Bird R, Roselli M, Hardman KD, Johnson S, Pope S, et al. In vivo tumor targeting of a recombinant single-chain antigen-binding protein. J Natl Cancer Inst 1990;82:1191-7.
- Kipriyanov SM. Generation of bispecific and tandem diabodies. Methods Mol Biol 2002;178:317-31.
- Kipriyanov SM, Moldenhauer G, Schuhmacher J, Cochlovius B, Von der Lieth CW, Matys ER, et al. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmco-kinetics. J Mol Biol 1999;293:41-56.
- Alt M, Muller R, Kontermann RE. Novel tetravalent and bispecific IgG-like antibody molecules combining single-chain diabodies with the immunoglobulin gamma1 Fc or CH3 region. FEBS Lett 1999;454:90-4.
- Lu D, Jimenez X, Zhang H, Atkins A, Brennan L, Balderes P, et al. Di-diabody: a novel tetravalent bispecific antibody molecule by design. J Immunol Methods 2003;279:219-32.
- Jung SH, Pastan I, Lee B. Design of interchain disulfide bonds in the framework region of the Fv fragment of the monoclonal antibody B3. Proteins 1994;19:35-47.
- FitzGerald K, Holliger P, Winter G. Improved tumour targeting by disulphide stabilized diabodies expressed in Pichia pastoris. Protein Eng 1997;10:1221-5.
- Worn A, Pluckthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol 2001;305:989-1010.
- Muyldermans S, Atarhouch T, Saldanha J, Barbosa JA, Hamers R. Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng 1994;7:1129-35.
- Riechmann L, Muyldermans S. Single domain antibodies: comparison of camel VH and camelised human VH domains. J Immunol Methods 1999;231:25-38.
- Els Conrath K, Lauwereys M, Wyns L, Muyldermans S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J Biol Chem 2001;276:7346-50.
- Miller K, Meng G, Liu J, Hurst A, Hsei V, Wong WL, et al. Design, construction, and in vitro analyses of multivalent antibodies. J Immunol 2003;170:4854-61.
- Lu D, Kotanides H, Jimenez X, Zhou Q, Persaud K, Bohlen P, et al. Acquired antagonistic activity of a bispecific diabody directed against two different epitopes on vascular endothelial growth factor receptor 2. J Immunol Methods 1999;230:159-71.
- Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Fab-scFv fusion protein: an efficient approach to production of bispecific antibody fragments. J Immunol Methods 2002;267:213-26.