
Role of norepinephrine in development of short-term myocardial hibernation
Introduction
Norepinephrine (NE) is the main transmitter released from sympathetic nerve terminals; it plays an important role in modulating the course of myocardial ischemia. Hibernation of the myocardium is defined as a condition of persistent myocardial dysfunction at rest due to reduced coronary blood flow, which can be partially or completely restored to normal by improving blood flow[1]. Hibernating myocardium constitutes the main part of recovery of heart function after reperfusion in patients with coronary heart disease, and is thought to be an important sign for revascularization in these patients[4]. Myocardial hibernation is thought to be an endogenous cardioprotective phenomenon characterized by adaptation to myocardial ischemia[1–3]; however, to date, the mechanisms responsible for the development and maintenance of hibernation remain unclear. The role of NE in the development of myocardial hibernation also remains unclear. In the present study, the role of NE in the development of short-term hibernation was investigated in isolated rat hearts. Myocardial infarction is often accompanied by an increase in NE in the myocardial and circulating system, which may play a role in the development of myocardial hibernation, so the effect of exogenous NE on the development of short-term myocardial hibernation was also investigated.
Materials and methods
Heart preparation Male Sprague-Dawley rats (200–250 g, 8 groups, n=6 rats/group) were pretreated with heparin (2 000 U/kg, ip), followed by sodium pentobarbitone (50 mg/kg, ip). After confirmation of complete anesthesia, the hearts were rapidly excised through a midline incision and immediately immersed in ice-cold modified Krebs-Henseleit buffer. Within 2 min, the hearts were perfused in a retrograde manner through the aorta, the vena cava and pulmonary veins were ligated, and the left ventricle was cannulated with a fluid-filled latex balloon through an incision in the left atrium. After the balloon was inserted into the left ventricle, the balloon was inflated to an end-diastolic pressure of about 6 mmHg (1 mmHg=0.133 kPa), and the left ventricular pressure from the balloon was measured by using low-compliance tubing, an MLT844 physiological pressure transducer, and an Octal Bridge amplifier (AD Instruments, Castle Hill, Australia). Hemodynamics were recorded on a Dell computer (Dell) using Chart software (v4.2.2, AD Instruments, Castle Hill, Australia) for Microsoft Windows. All hearts were paced at 300 beats per min throughout by using a pacing electrode secured to the right atrium. The basic perfusion fluid was a phosphate-free modified Krebs-Henseleit buffer containing 11 mmol/L glucose and having the following ionic composition[5] (in mmol/L): 144.0 Na+, 6.0 K+, 2.5 Ca2+, 130.0 Cl–, 1.2 SO42–, 25.0 HCO3–, and 0.5 Na2EDTA. The buffer was gassed with 95% O2, 5% CO2, was temperature controlled at 37 °C by 2 water baths, and the pH was adjusted to 7.4. The hearts were kept in a glass water-heated jacket, which kept the surrounding air temperature at 37°C. The basic perfusion pressure was 90 cmH2O, and the low-flow perfusion pressure was 15 cmH2O. Coronary flow was measured by using timed collections of the coronary effluent with graded glass cylinders throughout the protocol, and aliquots of perfusate were collected for lactate dehydrogenase (LDH) analysis. Samples for the estimation of LDH were collected in chilled tubes and stored at -20 °C until assayed by colorimetry.
All animals were provided by the Experimental Animal Center of Tongji College, Huazhong University of Science and Technology, and received humane care in compliance with the principles outlined in the Guide for the Care and Use of Laboratory Animals.
Protocol After equilibrium was reached in the 20-min perfusions, hearts were divided into 8 groups as follows: group I: hearts were perfused under a constant pressure of 90 cmH2O for 150 min; group II: hearts were switched to the low-flow ischemia mode, and reperfused for 30 min after 120 min of low-flow ischemia; group III: hearts were switched to low-flow ischemia and a dose of 1×10–6 mol/L dobutamine in 100 µL of water[5] was injected into a side arm immediately above the aortic cannula after 120 min of low-flow ischemia; group IV (NS group): 0.9% sodium chloride (7 mL/kg, ip, 24 h before experiment)-treated hearts were switched to the low-flow ischemia mode and after 120 min of low-flow ischemia, the apical part of each heart was cut off on ice and immediately put into fixation solution, and the remaining part was freeze-clamped and stored in liquid nitrogen for biochemical analysis; group V (reserpine group): reserpinized hearts (reserpine, 7 mg/kg, ip, 24 h before experiment; Guangdong Banmin Pharmaceutical Co) were studied as in group IV; group VI (NE group): hearts were studied as in group IV except for the presence of norepinephrine L-bitartrate (75 ng/mL, Sigma-Aldrich, St Louis, USA) in perfusion. Depletion of NE stores in the hearts of reserpinized rats was measured by testing the effect of tyramine (5 µg/mL, Sigma-Aldrich, St Louis, USA) in the perfusion buffer on the heart rates of spontaneously beating hearts (group VII)[6]. The same measurements were carried out for the hearts of non-reserpinized control animals (group VIII).
Myocardial ultrastructure examination The apical part of each heart was cut down on ice and immediately put into a 2.5% glutaraldehyde fixation solution at 4 °C. The subendocardium was subdivided into tissue blocks of 1 mm3, which were again put into the fixation solution for 30-min oscillation and fixation. Electron microscope slices were produced according to routine methods for the preparation of transmission electron microscope specimens. Ultrathin sections were stained with uranyl acetate and lead citrate for examination on a transmission electron microscope.
In situ terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling The left ventricle was immersion-fixed in 10% neutral formalin and embedded in paraffin. Serial sections of 3-µm thickness were produced. Apoptosis was detected by using the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) technique with an in situ cell death detection kit (AP, Roche, Germany) according to the manufacturer’s instructions. The numbers of TUNEL-positive cardiomyocytes were counted by 2 blinded observers using microscopes with an eyepiece grid (magnification, ×400) using the HPIAS-2 000 image analysis system. Five different visual fields were counted in each slice. The cardiomyocytes that had blue-stained nuclei were TUNEL-positive cardiomyocytes. TUNEL-positive nuclei that could not be definitively confirmed to have myocyte origins were excluded. The extent of apoptosis was expressed by normalizing the results to the number of myocyte nuclei per visual field in each sample.
In situ immunohistochemistry for Bcl-2 and Bax expression Each specimen was cut into 5-μm sections. The sections were deparaffinized with two 5-min changes of xylene and two 3-min changes of 100% alcohol. The slides were then washed in distilled water, and intrinsic peroxidase activity was inhibited with 3% hydrogen peroxide. After being rinsed, the slides were treated with 5% normal serum in 0.01 mol/L phosphate buffered saline for 10 min to block nonspecific binding. The sections were then incubated at room temperature overnight with monoclonal mouse anti-human Bcl-2 oncoprotein or monoclonal mouse anti-human Bax oncoprotein (Beijing Zhongshan Biotechnology Co) at dilutions of 1:50 (for both). The sections were then rinsed with phosphate buffered saline and incubated with biotin-conjugated goat antimouse IgG (Beijing Zhongshan Biotechnology Co) for 30 min. After being rinsed, the sections were incubated for 25 min, then after further rinsing, they were incubated for 30 min with a S-A/HRP work solution at 37 °C, then stained with diaminobenzidine (DAB) solution. Each section was photographed under a microscope with an eyepiece grid (magnification, ×400) using the HPIAS-2 000 image analysis system. Twenty randomly chosen visual fields for each group were used for computer-based automatic calculation of the mean absorbance of areas of immunopositive expression.
Biochemical analysis Adenosine 5'-triphosphate (ATP) concentration in myocardium was determined by using a bioluminescence method as described by Sperlagh and Krisztina et al[7,8]. A luminometric method based on the luciferin-luciferase reaction was used to quantify ATP in myocardium by using an ATP assay kit (Beyotime Bio-technology). The concentration of phosphocreatine in myocardium was determined by using the reverse phase-high performance liquid chromatography (RP-HPLC) methods described by Zhang et al[9]. The phosphocreatine standard was from Sigma-Aldrich. The concentration of glycogen in myocardium was determined with a chemistry colorimetry method by using a glycogen detection kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Detection of LDH LDH leakage from each heart was measured in the coronary effluent with a linked-enzyme spectrophotometric assay by using a lactate dehydrogenase kit (Zhongsheng Beikong Bio-technology and Science), as described by Bergmeyer[10]. LDH leakage was expressed as international units of LDH released per min per gram wet heart.
Data reduction and statistical analyses The pressure-rate product (PRP), which is the product of heart rate and left ventricular developed pressure, was calculated and used to assess the mechanical function, because it provides a method for quantification of the performance of a heart (for comparison with the same heart at a different time or with other hearts).
All data were expressed as mean±SD. Student’s paired or unpaired t-test was used to assess the differences between any 2 groups, and analysis of variance was used to assess differences among groups using SPSS 12.0 for Windows. P<0.05 was considered statistically significant.
Results
Recovery of cardiac function In group I, the hearts were perfused under a constant pressure of 90 cmH2O, and coronary flow, PRP, and LDH leakage were 11.9 mL/min, 23 236 mmHg·beat-1·min-1 and 64.25 mU·min-1·g-1, respectively,. There was no significant difference throughout the experiment. In group II, with the onset of ischemia, coronary flow, PRP, and LDH leakage promptly fell to 8.5%, 12.2%, and 23.7% of their pre-ischemic values, respectively, and there was no further significant decline over the next 2 h of low-flow ischemia. However, coronary flow and PRP promptly increased to control levels after reperfusion. In group III, hearts were strongly responsive to dobutamine: a bolus of dobutamine (1×10-6 mol/L) elicited a marked increase in systolic pressure (from 20.5 to 65.0 mmHg·beat-1·min-1, P<0.01) but a non-significant elevation in end-diastolic pressure (from 6.3 to 6.7 mmHg·beat-1·min-1, P>0.05).
There was no significant difference between the PRP values of the NS group and the reserpine group before ischemia, at the beginning of ischemia or after 120 min of ischemia. In the NE group, the PRP values were comparable to those of the other 2 groups after 20-min equilibrium perfusion, but the PRP values increased significantly after the addition of NE to the perfusion solution. With the onset of ischemia, the PRP values immediately fell to the same level as observed for the other 2 groups, then decreased progressively to 1005.1 mmHg·beat-1·min-1, which was significantly less than the comparable values in the other 2 groups (P<0.05) (Table 1).
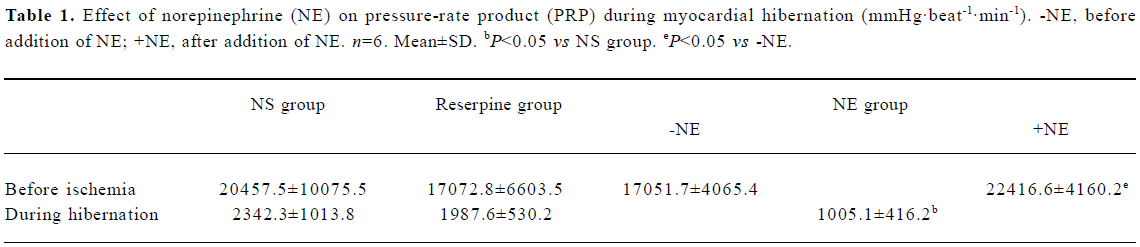
Full table
Tyramine test After addition of tyramine 5 µg /mL to the perfusion buffer, the heart rate of spontaneously beating hearts from non-reserpinized control rats was significantly enhanced from 308.2 to 409.8 beat/min (P<0.05), but the heart rate of spontaneously beating hearts from reserpinized rats was not significantly affected by tyramine (from 275.8 to 285.7 beat/min; P>0.05), which indicated that myocardial NE was depleted in reserpinized rats.
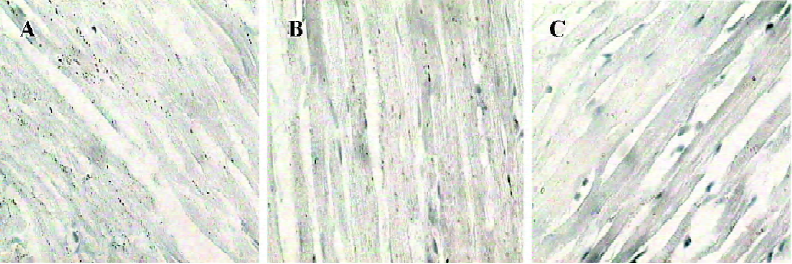
Myocardial ultrastructure Cardiac muscle fibers in the NS group and reserpine group were abundant, with regular arrays of myofibrils closely arranged within the sarcomere. The mitochondria were intact and had no swelling or disruption. The amount of glycogen in the mitochondria decreased, but cardiac muscle fibers and mitochondria from the NE group were significantly different from those in the other groups. The differences included a loss of myofibrillar content, myofibrillar disarray, and mitochondrial swelling, disruption and vacuolation. The glycogen content in the mitochondria decreased significantly (Figure 1, Table 3).
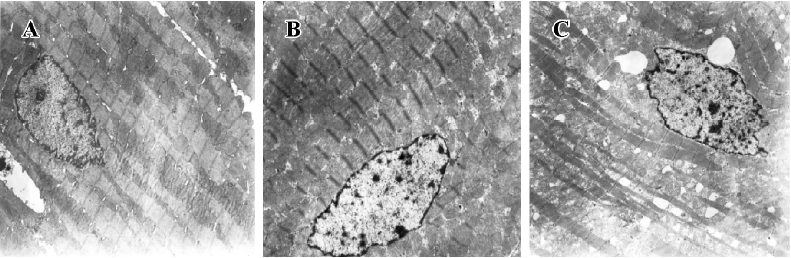
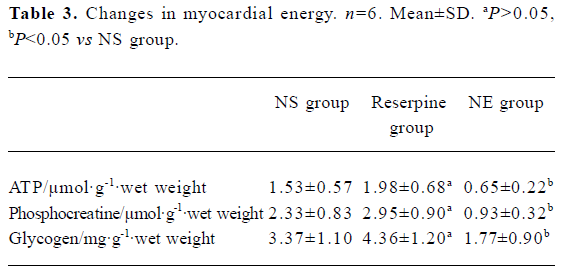
Full table
Injury to myocardial cell membranes LDH leakage was used to assess injury sustained by the myocardial cell membrane. The rate of LDH leakage was 14.8±3.0, 13.8±3.8, and 22.5±5.4 mU·min-1·g-1 after 120-min ischemia in the NS group, reserpine group, and NE group, respectively. There was no significant difference between the former 2 values, but compared with the NS group, the rate of LDH leakage was significantly higher in the NE group (P<0.05).
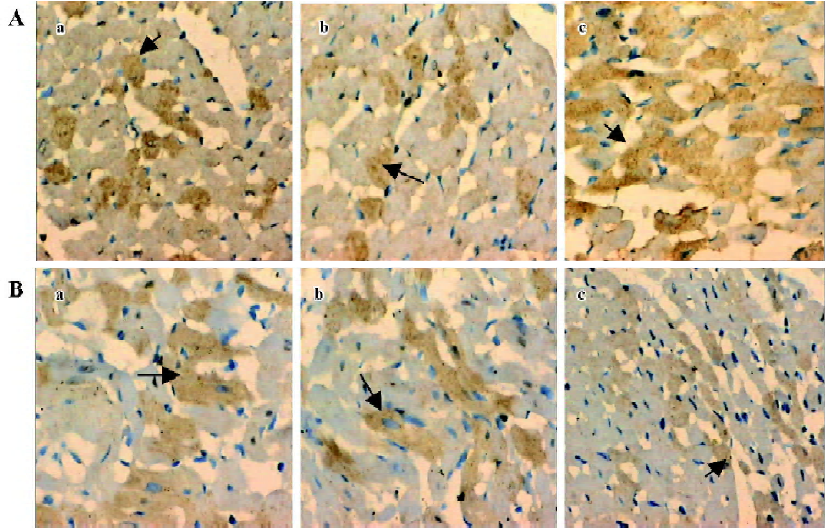
Analysis of myocyte apoptosis As assessed by TUNEL, there was no significant difference in the number of apoptotic myocytes between the NS group and the reserpine group, but the number of apoptotic myocytes was significantly greater in the NE group than in the NS group. The dark yellow immunoreactive Bcl-2 or Bax products were found in the cytoplasm of myocytes at various levels in the 3 groups. There was no significant difference between the NS group and reserpine group with respect to the mean absorbance of areas of Bcl-2 and Bax immunopositive expression, but the mean absorbance of areas of Bcl-2 and Bax immunopositive expression were significantly lower and higher, respectively, in the NE group than those in the NS group (Table 2, Figures 2,3).
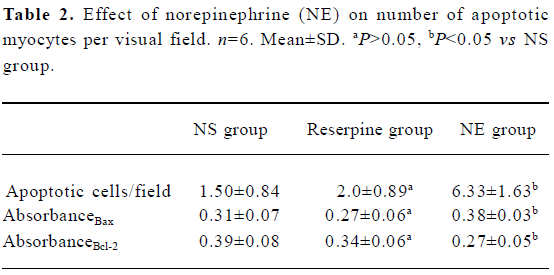
Full table
Changes in myocardial energy Compared with the NS group, there was more ATP, phosphocreatine and glycogen content in the reserpine group, but the difference was not significant (P>0.05). These values were significantly lower in the NE group compared to the other 2 groups (P<0.05, Table 3).
Changes in coronary flow There was no significant difference in coronary flow among the NS, reserpine, and NE groups before ischemia, but after 120 min ischemia, coronary flow was significantly lower in the NE group compared with the other 2 groups (between which the difference was not significant) (Table 4).

Full table
Discussion
Short-term myocardial hibernation model Myocardial hibernation is a term that was first used by Rahimtoola[1] to describe a postulated condition of chronic sustained abnormal contraction because of chronic underperfusion in patients with coronary heart disease, in whom revascularization or an improved oxygen supply-demand relationship could cause recovery of regional function. Initially, the concept of myocardial hibernation was based on clinical observations only, but this phenomenon has indeed been observed in experimental studies in vitro and in vivo. To describe this phenomenon, Ross introduced the term “perfusion-contraction matching”[11]. With reference to sustained perfusion-contraction matching, as demonstrated in experimental studies over several hours, Ross defined “short-term hibernation” and distinguished it from “chronic hibernation”, that is, a hypothetical condition of chronic perfusion-contraction matching. The existence and adaptive nature of short-term hibernation has been accepted, but the existence of perfusion-contraction matching over prolonged periods of time (chronic hibernation) has been questioned, and chronic hibernation has been suggested to be a result of repetitive or cumulative myocardial stunning[3,12,13].
In our perfusion pressure-controlled isolated rat heart model, low-flow ischemia induced sustained hypo-contraction, that is, sustained perfusion-contraction matching during which there was no morphological or biological evidence of myocardial injury: the PRP recovered to control levels after reperfusion, and hearts retained their responsiveness to dobutamine. These observations accord with those for short-term hibernating myocardium[5,14]. Our experiments were performed in isolated buffer-perfused rat hearts, a model that allows the function of myocardial NE during the development of short-term hibernation to be studied, and the degree and composition of coronary flow, heart rate, temperature, and myocardial function to be to precisely controlled in the absence of systemic neuroendocrine changes.
Role of myocardial NE in the development of short-term hibernation The observed reduction in myocardial contractile function during hibernation is not regarded as the consequence of a sustained energetic deficit, but instead as a regulatory event that serves to avoid an energetic deficit and to maintain myocardial integrity and viability[5,14]. In this active adaptation, activation of ATP-dependent potassium channels and increases in the concentration of interstitial adenosine are not involved in the development of short-term myocardial hibernation. In a study by Heusch et al it was suggested that a decrease in transient calcium and calcium responsiveness might contribute to the development of short-term hibernation[15]. Whereas the density of myocardial β adrenoceptors has been shown to be reduced in chronic animal models of myocardial hibernation[17] or stunning, β adrenoceptor density and affinity was unaltered in short-term hibernating myocardium after 90 min of moderate ischemia[16], which is consistent with our present results. In our short-term hibernation model in isolated rat heart, there was no significant difference between the NS group and reserpine group with respect to contraction function, coronary flow, ATP content, phosphocreatine content, glycogen content, myocardial ultrastructure, LDH leakage, apoptosis of myocytes according to TUNEL, or the amount Bax/Bcl-2 products, suggesting that myocardial NE may not contribute to the development of short-term hibernation, and that there might not be significant myocardial NE release during short-term hibernation, because significant NE release may injure myocytes[6,17,18].
Effect of exogenous NE on short-term hibernation Although short-term hibernation has been mimicked both in vitro and in vivo, there are many significant differences between them. Among these differences is the in vitro absence of neural or hormonal factors that have an important role in the regulation of cardiac performance and metabolic processes. NE is the main transmitter released from sympathetic nerve terminals and is an important component of blood. During myocardial infarction, the concentration of NE increases not only in the damaged myocardium but also in the circulating system because of increased peripheral releases from the pain, anxiety and reflex activation of the sympathetic nerve system. The excessive activation of the sympathetic nerve system is an important factor in accelerating ventricular remodeling and delayed heart failure. NE can increase cardiac performance, but it may also increase oxygen cost and apoptosis or injury to myocytes, decrease cardiac performance, injure the adaptation mechanism, and destroy the development of hibernation. In the present study, NE induced significant increases in cardiac performance despite producing no significant change in coronary flow during normal perfusion, but it induced significant decreases both in cardiac performance and coronary flow after 120 min ischemia, suggesting that the concentration of NE that induces increases in cardiac performance during normal perfusion induces decreases in cardiac performance during hibernation. ATP, phosphocreatine, and glycogen content was significantly lower in the NE group than in the NS group, suggesting that the balance of energy metabolism and perfusion-contraction matching was destroyed by high concentrations of NE.
Myocyte loss may occur in 2 distinct ways in the myocardium: apoptosis and necrosis. In our experiments, the proportion of apoptotic myocytes as observed by TUNEL was significantly greater in the NE group relative to the NS group, suggesting that a high concentration of NE might induce apoptosis of myocytes during hibernation, which could partially explain why NE induced the decrease in cardiac performance during hibernation. The bcl-2 proto-oncogene diagfamily is critical in the control of apoptosis. It has been proposed that cell viability after an apoptotic stimulus may depend on the ratio of Bcl-2 to Bax[19]. The mean absorbance of Bcl-2 immunopositive expression was significantly lower in the NE group than in the NS group, and the mean absorbance of Bax products was significantly higher in the NE group relative to the NS group, which indicates that NE increases the expression of Bax and decreases the expression of Bcl-2 during hibernation. The Bcl-2/Bax ratio became smaller, suggesting that the Bcl-2/Bax ratio may have an important role in the apoptosis of myocytes induced by NE during hibernation. It has been reported that NE induced the apoptosis of myocytes mainly by activating the β1 recep-tor[20], and mitogen-activated protein kinase and the reactive oxygen species mechanism may also have a role[21,22]. In the present study, NE not only induced the apoptosis of myocytes, but also induced injury to the myocardial cell membrane and caused changes in the ultrastructure of the myocardium, suggesting that NE may also induce necrosis in myocardium during hibernation. Another important reason why NE induces apoptosis and necrosis is that the decrease in coronary flow resulted in a decrease in energy supply to the myocardium, which might induce surviving myocytes to undergo apoptosis or necrosis[23].
The present study indicated that NE induced progressive decreases in coronary flow and cardiac performance, which might result from increases in apoptosis and necrosis, suggesting that NE may be an important factor in the deterioration of myocardial structure and function during hibernation, and that anti-adrenergic treatment may be helpful in the development and sustainment of short-term hibernation. However myocardial NE may not contribute to the development of short-term hibernation.
References
- Rahimtoola SH. A perspective on the three large multicenter randomized clinical trials of coronary bypass surgery for chronic stable angina. Circulation 1985;72 Suppl V:V-123-35.
- Arend FLS, Jeroen JB, Don P. Clinical assessment of myocardial hibernation. Heart 2005;91:111-7.
- Heusch G, Schulz R, Rahimtoola SH. Myocardial hibernation: a delicate balance. Am J Physiol Heart Circ Physiol 2005;288:H984-99.
- Westaby S. Coronary revascularization in ischemic cardiomyo-pathy. Surg Clin North Am 2004;84:179-99.
- Southworth R, Garlick PB. Dobutamine responsiveness, PET mismatch, and lack of necrosis in low-flow ischemia: is this hibernation in the isolated rat heart? Am J Physiol Circ Physiol 2003;285:H316-H324.
- Rump AFE, Klaus W. Evidence for noradrenaline cardiotoxicity mediated by superoxide anion radicals in isolated rabbit hearts. Naunyn Schmiedebergs Arch Pharmacol 1994;349:295-300.
- Sperlagh B, Kittel A, Lajtha A, Vizi ES. ATP acts as fast neurotransmitter in rat habenula: neurochemical and enzyme cytochemical evidence. Neuroscience 1995;66:915-20.
- Kordas KS, Sperlagh B, Tihanyi T, Topa L, Steward MC, Varga G, et al. ATP and ATPase secretion by exocrine pancreas in rat, guinea pig, and human. Pancreas 2004;29:53-60.
- Zhang YW, Long C, Ding LL. Determination of phosphocreatine in muscular tissues by high performance liquid chromato-graphy. Chin J Chromatogr 2001;19:251-2.
- Bergmeyer HU. Methods of enzymatic analysis. 3rd ed. Vol III. New York: Wiley; 1984. p 126–32
- Ross J Jr. Myocardial perfusion-contraction matching. Implications for coronary heart disease and hibernation. Circulation 1991;83:1076-83.
- Berry GJ, Masek M. The pathology of hibernating myocardium. Nucl Med Commun 2002;23:303-9.
- Shen YT, Vatner SF. Mechanism of impaired myocardial function during progressive coronary stenosis in conscious pigs. Hibernation versus stunning. Circ Res 1995;76:479-88.
- Heusch G. Hibernating myocardium. Physiol Rev 1998;78:1055-85.
- Heusch G, Rose J, Skyschally A, Post H, Schulz R. Calcium responsiveness in regional myocardial short-term hibernation and stunning in the in situ porcine heart: inotropic responses to postextrasystolic potentiation and intracoronary calcium. Circulation 1996;93:1556-66.
- Schulz R, Rose J, Martin C, Heusch G. Development of short-term myocardial hibernation: its limitation by the severity of ischemia and inotropic stimulation. Circulation 1993;88:684-95.
- Waldenstrom AP, Hjalmarson AH, Thornell L. A possible role of noradrenaline in the development of myocardial infarction. Am Heart J 1978;95:43-51.
- Rump AFE, Schierholz J, Klaus W. Studies on the cardiotoxicity of noradrenaline in isolated rabbit hearts. Arzneim-Forsch Drug Res 2002;52:543-51.
- Oltvai ZN, Korsmeyer SJ. Checkpoints of dueling dimers foil death wishes. Cell 1994;79:189-92.
- Ponicke K, Heinroth-Hoffmann I, Brodde OE. Role of beta 1- and beta 2-adrenoceptors in hypertrophic and apoptotic effects of noradrenaline and adrenaline in adult rat ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol 2003;367:592-9.
- Fu YC, Chi CS, Yin SC, Hwang B, Chiu YT, Hsu SL. Norepinephrine induces apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species−TNF alpha-caspase signaling pathway. Cardiovasc Res 2004;62:558-67.
- Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis. J Cell Physiol 2001;189:257-65.
- Depre C, Taegtmeyer H. Metabolic aspects of programmed cell survival and cell death in the heart. Cardiovasc Res 2000;45:538-48.