
Structure-based drug design of a novel family of chalcones as PPARα agonists: virtual screening, synthesis, and biological activities in vitro1
Introduction
Hypertriglyceridemia and hypercholesterolemia are 2 major risk factors for coronary heart disease (CHD)[1–3], which remains the leading cause of death in the developed countries. Fibrate drugs (Figure 1) have been widely used for the clinical treatment of dyslipidemia by lowering serum triglycerides and raising HDL cholesterol (HDLc), and remain the first choice for the treatment of severe hypertriglyceridemia[4–6]. Several studies have provided evidence that the hypolipidemic effect of fibrate drugs is attributed to the activation of peroxisome proliferator-activated receptors (PPAR)α[7,8], one of the 3 isoforms (α, γ, and δ) of the PPAR[9]. PPARα is highly expressed in the liver, heart, and muscle with a range of fatty acids in its natural ligands. In the human body, the activation of PPARα increases the clearance of the TG-rich, very low-density lipoprotein (VLDL) via the reduction of plasma levels of ApoCIII8[10–12] and the upregulation of ApoA1, which is the principal lipoprotein component of HDL[12–14]. The elevation of HDLc levels observed with fibrates arises partially due to the transcriptional induction of the major HDL apolipoproteins, apoA-I and apoA-II[15]. Regulating PPARα is also implicated as an anti-atherosclerosis approach[16]. Agonists have been shown to downregulate the expression of VCAM-1, to inhibit NF-κB and AP-1, and to mediate the reduction of plasma levels of interleukin-6, fibrinogen, and C-reactive protein[17].
Although fibrates are ligands for the PPARα receptor, they only show weak agonist activity as shown in Table 1[18,19]. More potent human PPARα agonists are expected to provide a better tool for studying the biology of PPARα and a superior clinical profile for therapeutic intervention in dyslipidemia and other metabolic disorders[20,21]. These reasons prompted us to discover novel small molecules as PPARα agonists with higher affinities.
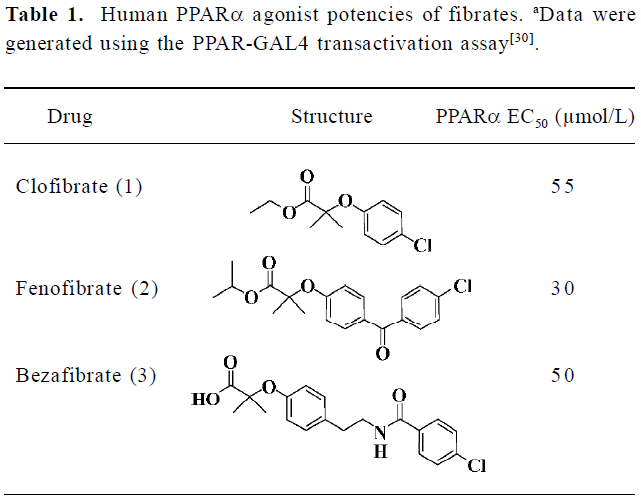
Full table
Chalcones, with a common 1,3-diphenyl-2-propen-1-one framework (Figure 1), have been known for over a century. Natural chalcones exist mainly as petal pigments and have also been found in the heartwood, bark, leaf, fruit, and root of a variety of trees and plants. Chalcone-containing plants, such as the Glycyrrhiza species have been used as folk remedies for a long time. Naturally occurring and synthetic chalcone compounds have shown interesting biological activities as shown by antioxidant, anti-inflammatory, anti-cancer, or anti-infective agents[22].
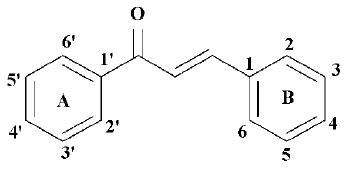
In this paper, we constructed a novel framework with the combination of the classical fibrate “head group” (Figure 2)[23], a linker with appropriate length and a chalcone. We designed 37 compounds and they were all identified by the structure-based virtual screening approach[21], based on the crystal structure of PPARα. According to the scores of virtual screening, structural similarity, and synthetic complexity, 6 compounds were selected, synthesized, and bioassayed; all of them showed high activation activities, with 50% effective concentration (EC50) values less than 2.5 µmol/L. Compounds 4d, 4e, and 4h are the most prominent, with EC50 values of 0.37 µmol/L, 0.63 µmol/L, and 0.06 µmol/L, respectively.

Materials and methods
Design of analogues of compounds for virtual screening In the first round of virtual screening, we designed compounds DE001-DE013 (Table 2) by keeping the structures of the fibrate “head group” (Figure 2) and chalcone, and changed the length and the structure of the linker. According to the result of the virtual screening (Table 2), we chose the ethoxy group (the linker of compound DE001) as the linker of our target compounds; the binding free energy of compound DE001 with PPARα is -9.18 kcal/mol, which is the most prominent among compounds DE001-DE013. In the second round of virtual screening, we designed compounds by keeping the fibrate “head group” and the ethoxy linker and changed the structures and substituents of the chalcones. By substituting position 3' of phenyl ring A of chalcone with the ethoxy linker and introducing electronic, hydrophobic, and steric bulky groups to phenyl ring B, we obtained compounds DE014-DE023; by substituting the 4' position of phenyl ring B of chalcone with the ethoxy linker and introducing electronic, hydrophobic, and steric bulky groups to phenyl ring A, we obtained compounds DE024-DE031; by introducing the ethoxy linker to positions 2' and 5' of phenyl ring A, we obtained compounds DE032-DE033; and by introducing the ethoxy linker to positions 3, 5, 2, and 6 of phenyl ring B, we obtained compounds DE034-DE037.
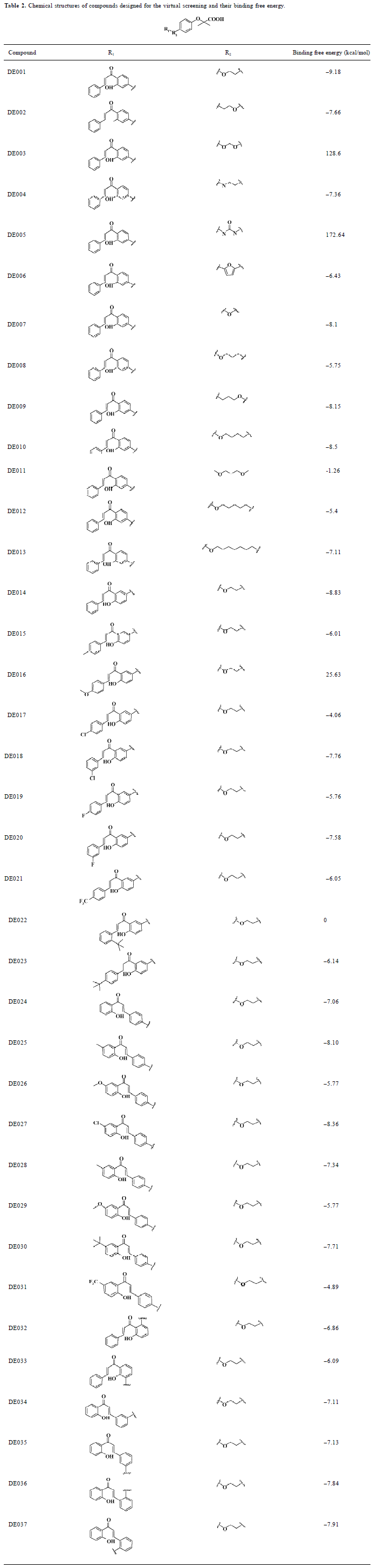
Full table
Virtual screening by molecular docking The crystal structure of PPARα complexing with GW409544 (1K7L)[23] recovered from the Brookhaven Protein Data Bank was used as the target for molecular docking. The docking calculations of compounds with PPARα were performed with the AutoDock 3.0 program (Morris et al, The Scripps Research Institute, La Jolla, CA, USA). Up to 20 different docking solutions were obtained for each molecule. All docking conformations in the different receptor conformers were then ranked with the AutoDock 3.0 scoring function[24]. For each compound, only the conformation with the highest score was chosen for analysis.
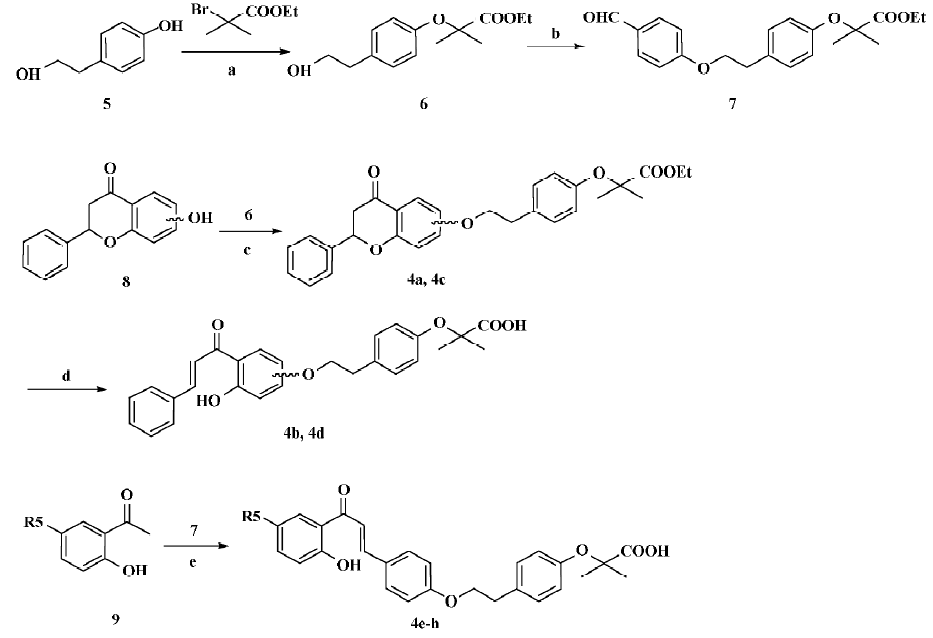
Synthetic procedures Figure 3 depicts the sequence of reactions that led to the preparation of compounds 4a–h. Compound 5 was converted to 6 by refluxing with 2-bromo-2-methyl-propionic acid ethyl ester in CH3CN in the presence of K2CO3[25], and 6 was condensed with p-hydroxy-benzaldehyde in tetrahydrofuran (THF) in the presence of PPh3 and diisopropyl azodicarboxylate (DIAD)(Mitsunobu reaction)[26], giving the key intermediate 7. Similarly, compound 8 was converted to 4a and 4c through the Mitsunobu reaction, then 4a and 4c were hydrolyzed using LiOH at room temperature to afford the target compounds 4b and 4d, respectively. Compound 9 was condensed with compound 7 by refluxing in CH3OH with 50% aq KOH, affording the target compounds 4e–h[27].
Biological assay The PPARα activation activities of the target compounds were monitored through published methods[28,29].
The response element (UASGAL4*5) was cloned upstream of the Pgl2-sv 40-Luc reporter (Promega, Madison, WI, USA), which contains the Simian virus early promoter for luciferase assay. GAL4 fusions were made by fusing human PPARc1 or PPARa ligand-binding domains (amino acids: 174–475) to the C-terminal end of the yeast GAL4 DNA-binding domain (amino acids: 1–147) of the pM1 vector. The pAdVAntage (Promega, Madison, WI, USA) vector was used to enhance luciferase expression.
HEK 293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2. At 1 d prior to transfection, the cells were plated to 50%–60% confluence in DMEM containing 10% delipidated FBS (DMEM-DFBS). The cells were transfected by Superfect (QIAGEN, Valencia, CA, USA) as per the manufacturer’s protocol. At 3 h after transfection, the reagent was removed and the cells were maintained in DMEM-DFBS. At 42 h after transfection, the cells were placed in phenol red-free DMEM-DFBS, treated for 24 h with the test compounds, and then collected with cell culture lysis buffer. Luciferase activity was monitored using the luciferase assay kit (Tropix, Bedford, MA, USA) according to the manufacturer’s instructions. Light emission was read in a Labsystems ascent fluoroskan reader (Flow Laboratories, Inc., Costa Mesa, CA, USA). To measure galactosidase activity to normalize the luciferase data, 50 µL supernatant from each transfection lysate was transferred to a new microplate. Galactosidase assays were performed in the microwell plates using a kit from Promega and read in a microplate reader. The agonist rates were calculated according to the activation data in 10 µmol/L compounds, and the EC50 values were estimated by fitting the activation data to a dose-dependent curve using a logistic derivative equation.
Results
Compound identification by virtual screening In order to avoid the synthesis of meaningless compounds and improve the efficiency of drug discovery, molecular modeling was employed to prescreen the designed compounds. Docking calculation was performed to rank the designed com-pounds. The binding free energy for each compound with PPARα is shown in Table 2. In most cases, the binding free energy between the designed compounds and PPARα shows a negative value less than -6 kcal/mol, indicating strong binding activity. As mentioned earlier, we chose the ethoxy group (the linker of compound DE001) as the linker for the target compounds according to the virtual screening results of compounds DE001-DE013 (Table 2). Similarly, compound DE014 is the most potent in this class, based on the virtual screening results of compounds DE014-DE023; compounds DE024-DE027 are the most potent and most representative among compounds DE024-DE031 according to the virtual screening result; and for compounds DE032-DE037, some also show excellent activities in terms of free energy scores. However, considering the binding free energy value, structural similarity, and synthetic complexity, we chose compounds DE001, DE014, and DE024-DE027 for further evaluation. As many drug candidates failed in clinical trials due to their poor ADMET properties, a prior consideration of their drug likeness would be of vital importance. The computational results of the drug likeness in Table 3 show that the 6 compounds meet most of the requirements in terms of the classic Lipinski rule-of-five. In addition, they satisfy 6 of 7 drug-like criteria developed by Zheng et al and Chen et al[30,31], with a high satisfaction rate of 0.9. As a result, the 6 compounds were finally chosen for synthesis and bioassay.
Full table
Analogues design and synthesis According to the results of the virtual screening, 6 compounds (4b, and 4d–4h) were synthesized; their chemical structures are shown in Table 4. These compounds were synthesized through the route outlined in Figure 3, and the details of synthetic procedures and structural characterizations are described in Appendix I.
Full table
Activation activity towards PPARα To determine the exact potency of the designed compounds, they were investigated in concentration-response studies and their EC50 values are also shown in Table 4. All showed EC50 values less than 2.5 µmol/L; 4d, 4e, and 4h are the most prominent, with EC50 values of 0.37 µmol/L, 0.63 µmol/L, and 0.06 µmol/L, respectively.
Discussion
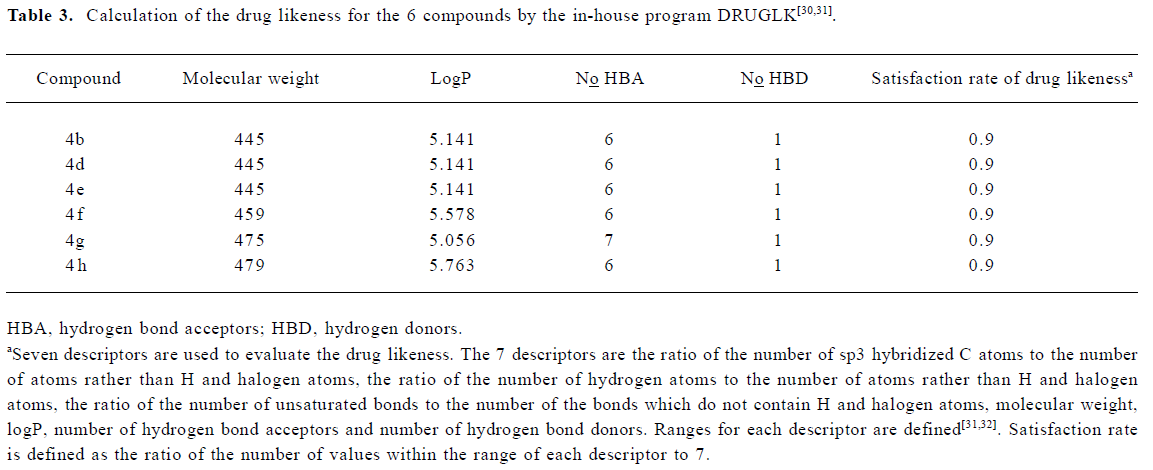
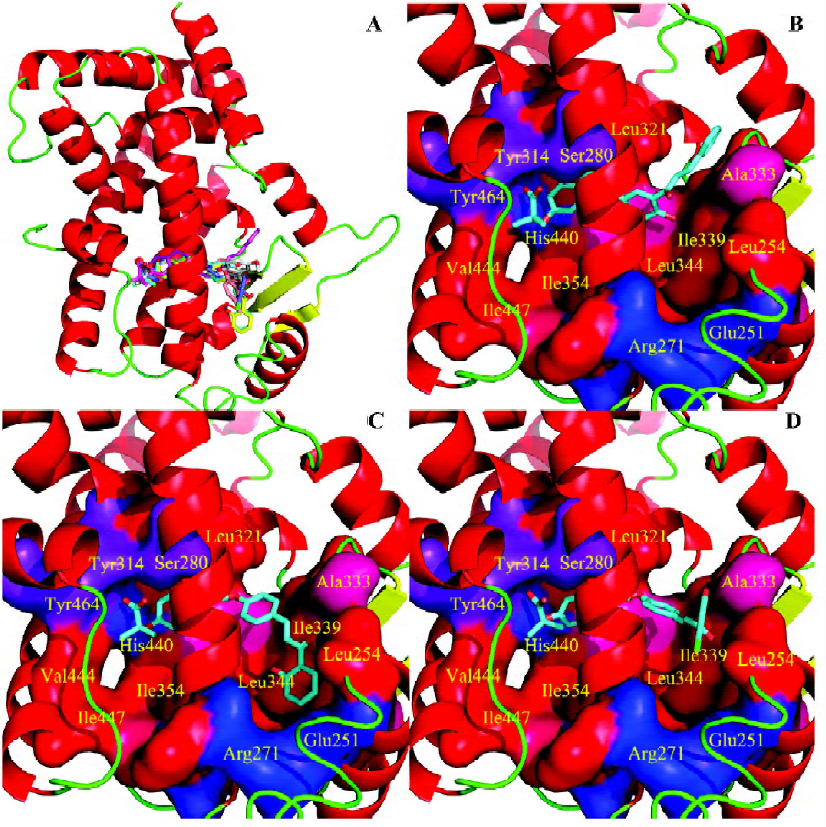
To gain structural information for further structural optimization, the 3-D binding models of the designed compounds to PPARα were generated based on the docking simulation. Figure 4A shows the superimposition of the 6 compounds within the binding pocket of PPARα. These binding models for the 6 compounds match that of the agonist GW409544 in the crystal structure[23] very well, especially the carboxyl head group. There is a strong electric field within the place where the head group locates, which forms strong electrostatic interactions to fix the head group, as shown from the polar motif of the surface in Figure 4 (4B–4D). However, the orientations of hydrophobic tails are different; they can be classified into 3 major conformations: tail up, tail down, and tail in the middle. The 3 most potent agonists 4d, 4e, and 4h represent the 3 major binding orientations of the hydrophobic tails, as shown in Figure 4 (4B–4D). This could be due to the fact that both the upper and lower arms of the “Y”-shaped pocket are hydrophobic, as shown from the hydropathicity surface in Figure 4 (4B–4D). Moreover, this result is consistent with the studies of Xu et al[23], who proved that the EPA in the crystal structure of PPARα also revealed the tail-up and tail-down configurations. Subsequently, we then mapped the detailed interaction (H-bonds and hydrophobic interaction) between the 3 most potent agonists 4d, 4e, and 4h and PPARα, as shown in Figure 5. Similarly, their head groups formed quite a few H-bonds with Ser280, Tyr314, His440, and Tyr464 in the binding pocket of PPARα, revealing a highly polar space under the AF-2 helix. Respectable hydrophobic interactions have been found between the tail of agonists and residues in the pocket, and the 3 agonists share some hydrophobic interactions with residues, such as Leu321, Val332, Phe273, and Cys276. However, some other hydrophobic interactions are different from each other because the orientations of the hydrophobic tails of the 3 agonists are different. For example, agonist 4d interacts with Met220, Leu331, and Met330 because its tail is in the upper arm of pocket while 4e does not, which may be due to its binding within the lower arm. This series of analogues interact with PPARα through a network of H-bonds involved the head acid group and numerous hydrophobic interactions by the tail.
In summary, 3 potent compounds (4d, 4e, and 4h) were discovered by using a structure-based virtual screening approach in conjunction with chemical synthesis and bioassay. We constructed a novel framework with the combination of the classical fibrate “head group” (Figure 2), a linker with appropriate length, and a chalcone. We designed 37 compounds on the basis of the novel framework; these compounds were identified by the structure-based virtual screening approach based on the crystal structure of PPARα. According to the scores of the virtual screening, structural similarity, and synthetic complexity, 6 compounds were chosen for synthesis and bioassay. Compounds 4d, 4e, and 4h were proven to be potent PPARα agonists, with EC50 values of 0.37 µmol/L, 0.63 µmol/L, and 0.06 µmol/L, respectively. To explore the binding characteristics of the designed compounds for further structural optimization, the binding models of compounds 4d, 4f, and 4i with PPARα were constructed based on molecular docking simulation. Their binding models are consistent with the binding model of agonist GW409544 with the crystal structure of PPARα, which demonstrates that our drug design strategy is reasonable and successful.
Appendix I
The reagents (chemicals) were purchased from Lancaster, Acros and Shanghai Chemical Reagent Company, and used without further purification. Analytical thin-layer chromatography (TLC) was HSGF 254 (150–200 µm thickness, Yantai Huiyou Company, China). Yields were not optimized. Melting points were measured in a capillary tube on a SGW X-4 melting point apparatus without correction. Nuclear magnetic resonance (NMR) spectra were performed on a Brucker AMX-300 NMR (IS as TMS). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low- and high-resolution mass spectra (HRMS) were given with electric (EI), electrospray, and matrix-assisted laser desorption ionization, produced by Finnigan MAT-95, LCQ-DECA spectrometer and IonSpec 4.7 Tesla.
2-(4-[2-Hydroxy-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (6) 4-(2-Hydroxy-ethyl)-phenol (6) (6.0 g, 43.2 mmol) was dissolved in CH3CN (100 mL), then 2-bromo-2-methyl-propionic acid ethyl ester (7.2 mL, 48.0 mmol) and K2CO3 (12 g, 87.0 mmol) were added in sequence. The reaction mixture was refluxed overnight, and K2CO3 was removed by filtration. Then the filtrate was condensed in vacuo and purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:4, v/v), to give 6 as a yellow oil. Yield: 45.9%. 1H NMR (CDCl3, 300 MHz): δ 1.25 (t, J=6.9 Hz, 3H), 1.57 (s, 6H), 2.79 (t, J=6.6 Hz, 2H), 3.80 (t, J=6.6 Hz, 2H), 4.22 (q, J=6.9 Hz, 2H), 6.79 (d, J=6.9 Hz, 2H), 7.08 (d, J=6.6 Hz, 2H); EI–MS m/z 252 (M+), 107 (100%); HRMS (EI) m/z calcd. C14H20O4 (M+) 252.1362, found 252.1357.
2-(4-[2-{4-Formyl-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (7) A solution of 2-(4-[2-hydroxy-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (6; 1.2 g 4.8 mmol) and p-hydroxybenzaldehyde (600 mg, 4.8 mmol) in THF (80 mL, redistilled from sodium) was treated at 0 °C with a preformed solution of PPh3 and DIAD, which was formed by the addition of DIAD (1.8 mL, 9.3 mmol) to a solution of PPh3 (2.5 g, 9.3 mmol) in THF (50 mL) at 0 °C. The reaction mixture was stirred at room temperature overnight, condensed in vacuo, purified by flash chromatography on silica gel, and eluted with a mixture of EtOAc/petroleum ether (1:8, v/v) to give 7 as a yellow oil. Yield: 29.7%. 1H NMR (CDCl3, 300 MHz): δ1.25 (t, J=7.5 Hz, 3H), 1.59 (s, 6H), 3.05 (t, J=7.2 Hz, 2H), 4.22 (m, 4H), 6.79 (d, J=6.6 Hz, 2H), 6.99 (d, J=9.0 Hz, 2H), 7.13 (d, J=8.7 Hz, 2H), 7.81 (d, J=6.9 Hz, 2H), 9.88 (s, 1H); EI-MS m/z 356 (M+), 121 (100%); HRMS (EI) m/z calcd. C21H24O5 (M+) 356.1624, found 356.1644.
2-Methyl-2-(4-[2-{4-oxo-2-phenyl-chroman-7-yloxy}- ethyl]-phenoxy)-propionic acid ethyl ester (4a) A solution of compound 6 (252 mg, 1.0 mmol) and 7-hydroxy-2-phenyl-chroman-4-one (240 mg, 1.0 mmol) in THF (30 mL, redistilled from sodium) was treated at 0 °C with a preformed solution of PPh3 and DIAD, which was formed by the addition of DIAD (400 µL, 2.0 mmol) to a solution of PPh3 (524 mg, 2.0 mmol) in THF (30 mL) at 0 °C. The reaction mixture was stirred at room temperature overnight, condensed in vacuo, purified by flash chromatography on silica gel, and eluted with a mixture of EtOAc/petroleum ether (1:8, v/v), to give 4a as a yellow oil. Yield: 55.3%. 1H NMR (CDCl3, 300 MHz): δ 1.25 (t, J=7.2 Hz, 3H), 1.62 (s, 6H), 2.98 (m, 4H), 4.16 (t, J=7.2 Hz, 2H), 4.24 (q, J=7.2 Hz, 2H), 5.45 (m, 1H), 6.49 (s, 1H), 6.59 (dd, J=9.0 Hz and 2.4 Hz, 1H), 6.78 (d, J=8.4 Hz, 2H), 7.14 (d, J=8.4 Hz, 2H), 7.40 (m, 5H), 7.84 (d, J=9.0 Hz, 1H); EI–MS m/z 474 (M+), 121 (100%); HRMS (EI) m/z calcd. C29H30O6 (M+) 474.2042, found 474.2034.
2-(4-[2-{3-Hydroxy-4-(3-phenyl-acryloyl)-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid (4b) Compound 4a (85 mg, 0.18 mmol) in a solvent of CH3OH (10 mL) and THF (10 mL) was added to 1 mol/L aqueous LiOH solution (2.5 mL, 2.5 mmol), and the mixture was stirred at room temperature for 6 h. CH3OH was removed in vacuo and water was added to the mixture. Then 10% HCl was added until pH=3. The mixture was then extracted with EtOAc (5 mL×3). The combined organic layer was dried, filtered, and condensed in vacuo to afford 4b as a yellow oil. Yield: 77.5%. 1H NMR (DMSO-d6, 300 MHz): δ 1.57 (s, 6H), 3.08 (t, J=6.6 Hz, 2H), 4.21 (t, J=6.6 Hz, 2H), 6.46 (m, 2H), 6.93 (d, J=8.4 Hz, 2H), 7.21 (d, J=8.4 Hz, 2H), 7.44 (m, 3H), 7.65 (m, 3H), 7.84 (m, 2H); EI-MS m/z 446 (M+), 121 (100%); HRMS (EI) m/z calcd. C27H26O6 (M+) 446.1729, found 446.1710.
2-Methyl-2-(4-[2-{4-oxo-2-phenyl-chroman-6-yloxy}-ethyl]-phenoxy)-propionic acid ethyl ester (4c) In the same manner as described in the preparation of 4a, 4c was prepared from 2-(4-[2-hydroxy-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (6) and 6-hydroxy-2-phenyl-chroman-4-one as a yellow oil. Yield: 49.8%; 1H NMR (CDCl3, 300 MHz): δ 1.18 (t, J=7.2 Hz, 3H), 1.49 (s, 6H), 2.84 (m, 4H), 4.03 (t, J=7.2 Hz, 2H), 4.12 (q, J=7.2 Hz, 2H), 5.56 (m, 1H), 6.72 (d, J=8.4 Hz, 2H), 7.04 (d, J=8.4 Hz, 1H), 7.20 (m, 4H), 7.42 (m, 3H), 7.51 (d, J=7.5 Hz, 2H); EI–MS m/z 474 (M+), 121 (100%); HRMS (EI) m/z calcd. C29H30O6 (M+) 474.2042, found 474.2027.
2-(4-[2-{4-Hydroxy-3-(3-phenyl-acryloyl)-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid (4d) In the same manner as described in the preparation of 4b, 4d was prepared from compound 4c as a yellow oil. Yield: 75.8%; 1H NMR (DMSO-d6, 300 MHz): δ 1.60 (s, 6H), 3.05 (t, J=6.9 Hz, 2H), 4.14 (m, 2H), 6.92 (m, 2H), 6.99 (m, 1H), 7.10 (m, 1H), 7.21 (m, 2H), 7.26 (m, 1H), 7.42 (m, 3H) 7.53 (m, 1H) 7.65 (m, 2H) 7.89 (m, 1H); EI–MS m/z 446 (M+), 121 (100%); HRMS (EI) m/z calcd. C27H26O6 (M+) 446.1729, found 446.1742.
2-(4-[2-{4-(3-[2-Hydroxy-phenyl]-3-oxo-propenyl)-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid (4e) 1-(2-hydroxy-phenyl)-ethanone (56 mg, 0.41 mmol) and 2-(4-[2-{4-formyl-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (7; 158 mg, 0.44 mmol) was dissolved in CH3OH (15 mL). To this solution, H2O (1 mL) and 50% aq. KOH (1 mL) were added successively, and the mixture was refluxed for 1 h. After cooling, the solution was poured onto crushed ice and acidified with 6 mol/L HCl. Then the mixture was extracted with EtOAc (5 mL×3). The combined organic layer was dried, filtered, and condensed in vacuo. Then the residue was purified by preparative TLC and eluted with a mixture of CH3OH/acetone/petroleum ether (1:30:30, v/v) to give 4e as a yellow oil. Yield: 3.8%. 1H NMR (DMSO-d6, 300 MHz): δ 1.60 (s, 6H), 3.07 (t, J=6.6 Hz, 2H), 4.20 (t, J=6.6 Hz, 2H), 7.00 (m, 6H), 7.21 (m, 2H), 7.51 (m, 4H), 7.91 (m, 2H); EI–MS m/z 446 (M+), 121 (100%); HRMS (EI) m/z calcd. C27H26O6 (M+) 446.1729, found 446.1740.
2-[4-(2-{4-[3-(2-Hydroxy-5-methyl-phenyl)-3-oxo-propenyl]-phenoxy}-ethyl)-phenoxy]-2-methyl-propionic acid (4f) In the same manner as described in the preparation of 4e, 4f was prepared from 1-(2-hydroxy-5-methyl-phenyl)-ethanone and 2-(4-[2-{4-formyl-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (7) as a yellow oil. Yield: 11.0%; 1H NMR (DMSO-d6, 300 MHz): δ 1.58 (s, 6H), 2.31 (s, 3H), 3.06 (t, J=6.9 Hz, 2H), 4.18 (t, J=6.9 Hz, 2H), 6.90 (m, 5H), 7.16 (d, J=8.4 Hz, 2H), 7.28 (m, 1H), 7.59 (m, 4H), 7.84 (m, 1H); EI–MS m/z 460 (M+), 298 (100%); HRMS (EI) m/z calcd. C28H28O6 (M+) 460.1886, found 460.1893.
2-[4-(2-{4-[3-(2-Hydroxy-5-methoxy-phenyl)-3-oxo-propenyl]-phenoxy}-ethyl)-phenoxy]-2-methyl-propionic acid (4g) In the same manner as described in the preparation of 4e, 4g was prepared from 1-(2-hydroxy-5-methoxy-phenyl)-ethanone and 2-(4-[2-{4-formyl-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (7) as a yellow oil. Yield: 34.6%; 1H NMR (DMSO-d6, 300 MHz): δ 1.59 (s, 6H), 3.06 (t, J =6.6 Hz, 2H), 3.84 (s, 3H), 4.21 (t, J=6.6 Hz, 2H), 6.96 (m, 5H), 7.11 (m, 3H), 7.35 (m, 1H), 7.43 (m, 1H), 7.58 (d, J=8.7 Hz, 2H), 7.82 (m, 1H); EI–MS m/z 476 (M+), 121 (100%); HRMS (EI) m/z calcd. C28H28O7 (M+) 476.1835, found 476.1836.
2-[4-(2-{4-[3-(5-Chloro-2-hydroxy-phenyl)-3-oxo-propenyl]-phenoxy}-ethyl)-phenoxy]-2-methyl-propionic acid (4h) In the same manner as described in the preparation of 4e, 4h was prepared from 1-(5-chloro-2-hydroxy-phenyl)-ethanone and 2-(4-[2-{4-formyl-phenoxy}-ethyl]-phenoxy)-2-methyl-propionic acid ethyl ester (7) as a yellow oil. Yield: 21.6%; 1H NMR (DMSO-d6, 300 MHz): δ 1.59 (s, 6H), 3.05 (t, J=6.9 Hz, 2H), 4.20 (t, J=6.9 Hz, 2H), 6.92 (m, 5H), 7.21 (d, J=8.4 Hz, 2H), 7.43 (m, 2H), 7.60 (d, J=9.0 Hz, 2H), 7.85 (m, 2H); EI-MS m/z 480 (M+), 121 (100%); HRMS (EI) m/z calcd. C27H25ClO6 (M+) 480.1340, found 480.1335.
References
- Anum EA, Adera T. Hypercholesterolemia and coronary heart disease in the elderly: a meta-analysis. Ann Epidemiol 2004;14:705-21.
- Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, et al. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes 1997;46:1319-27.
- Banner CD, Gottlicher M, Widmark E, Sjovall J, Rafter JJ, Gustafsson JA. A systematic analytical chemistry/cell assay approach to isolate activators of orphan nuclear receptors from biological extracts: characterization of peroxisome proliferator-activated receptor activators in plasma. J Lipid Res 1993;34:1583-91.
- Chong PH, Bachenheimer BS. Current, new and future treatments in dyslipidaemia and atherosclerosis. Drugs 2000;60:55-93.
- Brown PJ, Winegar DA, Plunket KD, Moore LB, Lewis MC, Wilson JG, et al. A ureido-thioisobutyric acid (GW9578) is a subtype-selective PPARalpha agonist with potent lipid-lowering activity. J Med Chem 1999;42:3785-8.
- Bunnelle WH, Dart MJ, Schrimpf MR. Design of ligands for the nicotinic acetylcholine receptors: the quest for selectivity. Curr Top Med Chem 2004;4:299-334.
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998;98:2088-93.
- El-Jack AK, Hamm JK, Pilch PF, Farmer SR. Reconstitution of insulin-sensitive glucose transport in fibroblasts requires expression of both PPARgamma and C/EBPalpha. J Biol Chem 1999;274:7946-51.
- Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem 2000;43:527-50.
- Hertz R, Bishara-Shieban J, Bar-Tana J. Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J Biol Chem 1995; 270: 13 470–5.
- Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992;358:771-4.
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 1995; 270: 12 953–6.
- Berthou L, Duverger N, Emmanuel F, Langouet S, Auwerx J, Guillouzo A, et al. Opposite regulation of human versus mouse apolipoprotein A-I by fibrates in human apolipoprotein A-I transgenic mice. J Clin Invest 1996;97:2408-16.
- Li Z, Liao C, Ko BC, Shan S, Tong EH, Yin Z, et al. Design, synthesis, and evaluation of a new class of noncyclic 1,3-dicarbonyl compounds as PPARalpha selective activators. Bioorg Med Chem Lett 2004;14:3507-11.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835-9.
- Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998;395:137-43.
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, . Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem 1999; 274: 32 048–54.
- Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA 1997;94:4312-7.
- Pinelli A, Godio C, Laghezza A, Mitro N, Fracchiolla G, Tortorella V, et al. Synthesis, biological evaluation, and molecular modeling investigation of new chiral fibrates with PPARalpha and PPARgamma agonist activity. J Med Chem 2005;48:5509-19.
- Wahli W, Braissant O, Desvergne B. Peroxisome proliferator activated receptors: transcriptional regulators of adipogenesis, lipid metabolism and more. Chem Biol 1995;2:261-6.
- Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA 2000;97:7124-9.
- Ni LM, Meng CQ, Sikorski JA. Recent advances in therapeutic chalcones. Exp Opin Ther Patents 2004; 14: 1669–91.
- Sierra ML, Beneton V, Boullay AB, Boyer T, Brewster AG, Donche F, et al. Substituted 2-[(4-aminomethyl)phenoxy]-2-methylpro-pionic acid PPARalpha agonists. 1. Discovery of a novel series of potent HDLc raising agents. J Med Chem 2007;50:685-95.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639-62.
- Tanaka N, Tamai T, Mukaiyama H, Hirabayashi A, Muranaka H, Ishikawa T, et al. Beta(3)-adrenoceptor agonists for the treatment of frequent urination and urinary incontinence: 2-[4-(2-[[(1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-methylethyl]amino]ethyl) phenoxy]-2-methylpropionic acid. Bioorg Med Chem 2001;9:3265-71.
- Ahn C, Correia R, DeShong P. Mechanistic study of the Mitsunobu reaction. J Org Chem 2002;67:1751-3.
- Daskiewicz JB, Depeint F, Viornery L, Bayet C, Comte-Sarrazin G, Comte G, et al. Effects of flavonoids on cell proliferation and caspase activation in a human colonic cell line HT29: an SAR study. J Med Chem 2005;48:2790-804.
- Park H, Lee J, Lee S. Critical assessment of the automated AutoDock as a new docking tool for virtual screening. Proteins 2006;65:549-54.
- Marcus SL, Miyata KS, Rachubinski RA, Capone JP. Transactiva-tion by PPAR/RXR heterodimers in yeast is potentiated by exogenous fatty acid via a pathway requiring intact peroxisomes. Gene Expr 1995;4:227-39.
- Henke BR, Blanchard SG, Brackeen MF, Brown KK, Cobb JE, Collins JL, et al. N-(2-Benzoylphenyl)-L-tyrosine PPARα agonists. 1. Discovery of a novel series of potent antihypergly-cemic and antihyperlipidemic agents. J Med Chem 1998;41:5020-36.
- Zheng SX, Luo XM, Chen G, Zhu WL, Shen JH, Chen KX, et al. A new rapid and effective chemistry space filter in recognizing a druglike database. J Chem Inf Model 2005;45:856-62.
- Chen G. Structure-based combinatorial focus library design: Metho-dologies and applications. [PhD Thesis]. Shanghai: Shanghai Institute of Materia Medica; 2003.