
Hepatotoxicity of anti-inflammatory and analgesic drugs: ultrastructural aspects1
Introduction
In recent years, the number of anti-inflammatory and analgesic drugs in use has increased. Toxic effects are occasionally reported, but only after a drug has been used for a significant period of time. Whether they are administered at a therapeutic dose or at higher levels, anti-inflammatory drugs and analgesics can produce various clinical, biochemical and structural changes. Frequently observed and extensively studied are the effects of nonsteroidal anti-inflammatory drugs (NSAIDs) and other analgesics on the liver.
Clinicians sometimes forget that reactions to drugs and xenobiotics can cause a whole range of liver diseases; the final diagnosis of liver disease should be based on clinical drug history, and laboratory and morphological findings. The importance of etiological diagnosis cannot be over-emphasized: withdrawal, over time, of the toxic drug can be followed by improvement or resolution of the liver damage.
Electron microscopy is an important step in detecting pharmacological and toxicological effects in human liver biopsies. According to Phillips et al, electron microscopy is “of equal or greater value than light microscopy” in the evaluation of patients with known or suspected drug- or toxin-induced liver damage[1]. Although light microscopy is certainly important in the diagnosis of hepatotoxicity, a number of subcellular alterations can only be identified by electron microscopy. The present review is focused on the contribution of electron microscopy in studies of the hepatotoxicity induced by a number of frequently used analgesic and anti-inflammatory drugs.
Toxic effects at the subcellular level: general considerations
The modalities of ultrastructural reactions to noxious agents are few. Mostly, the changes observed depend on the chemical or physical nature, composition, and concentration and duration of exposure to the damaging agent. Despite their different origins, histological features and functions, cells in general and liver cells in particular tend to show a limited number of changes in membranes, organelles and nuclei in response to noxious agents. Succinct descriptions can contribute to an understanding of the more specific subcellular damage caused by various drugs.
The acinus concept, first proposed by Rappaport in 1969[2], explained the zonal distribution of damage according to the blood supply of the acinus, the smallest functional unit within the liver. Oxygen tension and nutrients in the sinusoid decrease in a gradient from the peripheral portal space (zone 1) to the center of the acinus, the terminal hepatic venule (perivenular zone 3). Electron microscopic examination confirms the localization of changes according to this model; that is, cells of zone 1 are the first to receive blood and nutrients, and usually the last to die and the first to regenerate. Alternatively, more distant cells may receive blood with less oxygen and nutrients, but also with less toxic agent, and thus may be more resistant to damage. Moreover, variations in the location of hepatotoxicity may be related to the gradient of blood supply and presence of the enzyme system responsible for the conversion of the drug into a toxic metabolite. For example, in overdose, acetaminophen becomes an intrinsic hepatotoxin that can cause perivenular necrosis. Thus, depending on the mechanism of toxicity and the drug involved, the acinar unit concept may explain the variability of findings not only in different liver specimens, but also within a single section of the same specimen.
Acute toxic cell injury, manifested by acute cell swelling and disintegration of organelles[3,4], is now rarely the result of medication toxicity and therefore is beyond the scope of this review. In less aggressive injury, drugs are metabolized in the liver by various reactions, including oxidation, reduction, hydrolysis and conjugation, and thus fat-soluble compounds are converted to hydrophilic ones, facilitating excretion into the urine. Hepatocytes are endowed with protective mechanisms against toxicity: (1) the oxidizing enzymes of the smooth endoplasmic reticulum (SER); (2) the glutathione oxidation–reduction system; (3) the peroxisomal H2O2 system; and (4) various cytosolic enzymes[5]. Only the organelles of hepatocytes that are primarily affected by drug damage will be mentioned here.
The endoplasmic reticulum is consistently altered during toxicity. In the early stages of toxicity, proliferation/reduplication of the SER can be observed. With ongoing toxicity, the SER comes to occupy most of the cell, both by proliferation and dilatation of its cisternae. Hepatic clearance of many toxins requires enhanced synthesis of the enzyme UDP-glucuronyl transferase by the SER. Pompella and Comporti, using confocal laser scanning fluorescence microscopy plus image video analysis, show that the perinuclear SER is the first to be involved in oxidative stress and lipid peroxidation[6]. The rough endoplasmic reticulum (RER), the major site of protein synthesis, is also the primary site of damage by hepatotoxins. Electron microscopic examination frequently reveals the detachment of ribosomes (“degranula-tion” of RER), which is considered to limit protein synthesis.
Mitochondria are extremely sensitive to drugs, both at normal dosages and especially in overdose. Ultrastructural changes have been classified as “reversible” and “irreversible”[4]. Anoxia has been implicated in mitochondrial changes, which explains the more severe findings in organelles located in acinar zone 3, away from the better oxygenated zone 1 (periportal). Mitochondrial components such as superoxide dismutase[7] and NADP-dependent dehydrogenase[8] have been localized by electron microscopy and can be used to analyze toxic injury. Mitochondria can be selectively injured by toxins that interfere with oxidative phosphorylation or electron transport. Mitochondrial function is inhibited by certain drugs through their effect on beta-oxidation energy production. In this way, nicotinamide adenine dinucleotide and flavin adenine dinucleotide synthesis is inhibited, resulting in decreased ATP production. The elevation of the serum transaminase[alanine transaminase (ALT) and aspartate transaminase (AST)] found in drug-induced liver diseases (DILD) has been linked to the association of this enzyme with the outer parts of the inner mitochondrial membrane[9].
Intramitochondrial dense granules (also named matrix granules or native granules) are osmiophilic and usually measure 20–50 nm in diameter. They are considered to have a role in mitochondrial calcium metabolism, and it is important to note that their frequency can be altered by drugs. Manov et al have shown that they disappear from cultured hepatoma-derived cells after cells are exposed to high doses of acetaminophen[10]. In their early description of the mechanism of hepatocyte injury and cell death, Desmet and Vos describe the disappearance of matrix granules as an early, reversible phenomenon (stage 1a)[4].
Flocculent or “wooly” densities in mitochondria are completely different from matrical granules: they appear later in the injury cascade, and are the most reliable early manifestation of irreversible injury, cell death and ensuing necrosis[11]. Ischemia/anoxia and a multitude of drugs and heavy metals, in addition to immune cytolysis[12] can generate the wooly densities. However, most frequently they are found in specimens undergoing delayed fixation or those that are retrieved post-mortem or from comatose patients or experimental animals.
Close association of mitochondria and endoplasmic reticulum[13] is seen in normal cells, but can be extensive under certain conditions. Likewise, drugs and hormones that alter mitochondrial metabolism have a deleterious effect on the RER[14]. (More specific mitochondrial changes, related to drugs, are listed in the following section and in Table 1).
There is little information concerning the effects of anti-inflammatory drugs and analgesics on peroxisomes. These organelles contain catalases, oxidases and carrier proteins[15]. Recently it has been shown that pretreatment with peroxisome proliferators protect mice against acute acetaminophen toxicity, but not other hepatotoxins[16]. Except for hyperplasia of hepatocytes, and an increase in peroxisomes and endoplasmic reticulum, peroxisome proliferators do not produce specific ultrastructural changes.
Golgi complexes of hepatocytes are frequently affected by toxins, thus producing widespread effects throughout the cells. Poisoning of the Golgi complex of rat liver cells with tris (hydroxymethyl)-amino-methane causes vesiculation of Golgi stacks on the concave side and swelling of lysosomes and secretory vesicles[17]. Major toxic changes can be observed in the nuclei and plasma membranes of hepatocytes. In liver biopsies, nuclei of individual cells can show evidence of apoptosis (heterochromatin margination clumping and nuclear fragmentation). Apoptosis may be initiated through the stimulation of death receptors located on the cell surface or through an intrinsic pathway involving the release of apoptotic signals from mitochondria[18]. Both signals converge on a cascade of cysteine proteases known as caspases, which are central to the initiation and execution of apoptosis. In cultured cells, nuclear changes are more conspicuous and are associated with other changes (see later in this review). The plasma membranes of hepatocytes may show interruption and disintegration in the necrotic foci. Covalent binding of the drug to intracellular proteins can cause a decrease in ATP levels, leading to actin disruption. Disassembly of actin fibrils at the surface of the hepatocyte causes blebs and rupture of the membrane[10,19]. Kupffer cells and sinusoidal lining cells may harbor prominent phagolysosomes, indicating uptake of remnants of dead cells. There is no drug-specificity in this finding, which is prominent in chronic, rather than acute toxicities. Electron microscopy can be used to identify stellate cells (Ito cells) in various stages of their transformation into fibroblasts, indicating potential fibrosis. Lysosomes in liver cells play an important role in drug metabolism and elimination of drugs. Typical lysosomotropic agents include gold-salts, methotrexate, and also acetaminophen and its metabolites. A number of experimental studies of drug-induced lysosomal disorders of the liver in man and laboratory animals were reviewed by Schneider et al. and show that many hepatotoxic drugs are in fact lysosomotropic agents[20].
Mechanisms of drug-induced liver diseases
Drugs causing DILD can be classified in various ways on the basis of the mechanisms by which they act. The classical divisions are: (1) drugs directly affecting the liver; and (2) drugs that mediate an immune response. The first group includes drugs that cause “predictable intrinsic” reactions; these are immediate, dose-related and reproducible in animals (eg acetaminophen). The second group, comprising drugs that cause “idiosyncratic” reactions, act through hypersensitivity (immunoallergy) and metabolic-idiosyncratic reactions [eg phenytoin, isoniazid (INH)]. These latter reactions are not predictable, are dose-independent and cannot be reproduced in experimental animals. Moreover, they appear after a period of latency lasting weeks to years[21]. Anti-inflammatory/analgesic drugs can be classified as follows:(1)intrinsic: aspirin, acetaminophen, phenylbutazone; (2) immunoallergic idiosyncratic: ibuprofen, sulindac, phenylbutazone, piroxicam, diclofenac; and (3) metabolic idiosyncratic: benoxaprofen, diclofenac, indomethacin, naproxen. It has been argued that these classifications are not valid because some drugs may exhibit characteristics of both major groups: they may show initially immediate, dose-related toxicity, only to produce, with ongoing use at lower dosages, signs of immune reaction (acetaminophen is suspected to act in this way).
For drugs in which hepatotoxicity is considered to be due to idiosyncratic reactions, abnormal immune mechanisms have been implicated. Covalent binding of a drug to the P-450 enzyme acts as an immunogen, activating cytolytic T cells and cytokines and stimulating a multifaceted immune response. Recently it has been suggested that drug hepatotoxicity could be classified as either hepatocellular or cholestatic toxicity[22]. Drugs that affect transport proteins at the canalicular membrane can interrupt bile flow. Loss of villous processes and interruption of transport pumps, such as multidrug resistance-associated protein 3, prevent the excretion of bilirubin, causing cholestasis. Moreover, toxic metabolites excreted in bile may cause injury to the bile duct epithelium.
Hepatotoxicity of specific drugs
(Gluco)corticosteroids The classical effect of prolonged corticosteroid therapy, both in children and adults, is steatosis[1,23]. Lipid droplets can be visualized with an optical microscope provided an adequate histological technique is used. Preliminary processing for electron microscopy, in which semi-thin sections of epoxy-embedded tissue are produced, is ideal for visualizing fat droplets stained with toluidine blue 1% solution: the fat droplets appear yellow or green against the blue coloration of the slides. In contrast, carbohydrate-containing compounds (ie, glycogen and mucopolysaccharides) exhibit metachromasia with red or pink coloring. Electron microscopy unmistakably identifies the multiparticulate α-glycogen molecules in hepatocytes thanks to their “flower” appearance. This is in contrast to the β-glycogen monoparticulate particles present in other cell types.
A rarely recognized side-effect of glucocorticosteroid therapy is liver enlargement. This has been noticed in children treated with large doses of prednisone for a variety of conditions [rheumatic fever, rheumatoid arthritis, bronchial asthma, nephritic syndrome, systemic lupus erythematosus (SLE), idiopathic thrombocytopenic purpura, Stevens-Johnson syndrome, aplastic anemia, hemolytic anemia, infantile spasms, and giant cavernous hemangioma][24]. During our investigation of prednisone-treated children, we found hepatomegaly in 7 of 122 patients treated with 2 mg/kg and 12 of 18 treated with 4 mg/kg prednisone. With the cessation of steroid therapy, liver size decreased within 7–14 d. One micrometer-thick sections stained with toluidine blue showed metachromasia of hepatocyte cytoplasm and a few large lipid droplets in these cases. Electron microscopy revealed that the hepatocyte cytoplasm was filled with glycogen, which displaced the mitochondria, RER and other organelles towards the plasma membrane or around the nucleus. The appearance of the hepatocytes was similar to that observed in some glycogen storage diseases or untreated diabetes mellitus.
Corticosteroids are also termed glucocorticosteroids because of their distinct effects on carbohydrate metabolism (ie gluconeogenesis, promotion of liver glycogen deposition, and elevation of blood glucose concentrations). Several mechanisms are considered to be involved in glucocorticoid-induced glycogen deposition: (1) glucocorticoids activate glycogen synthetase a through an indirect mechanism involving synthesis of a phosphatase; (2) following steroid-induced hyperglycemia, there is increased glycogen deposition through activation of phosphorylase a by glucose and subsequent activation of synthetase a; and (3) glycogen synthetase is activated by insulin, which increases in glucocorticoid-induced hyperglycemia[24,25].
Other noteworthy ultrastructural effects of corticosteroids are: (1) enlargement of mitochondria; (2) increase in frequency of lysosomes; (3) decrease in frequency of peroxisomes; (4) increased volume of hepatocytic cytoplasm, but decreased area of RER and SER following the accumulation of other cytosolic components (mainly fat droplets and glycogen)[1,24].
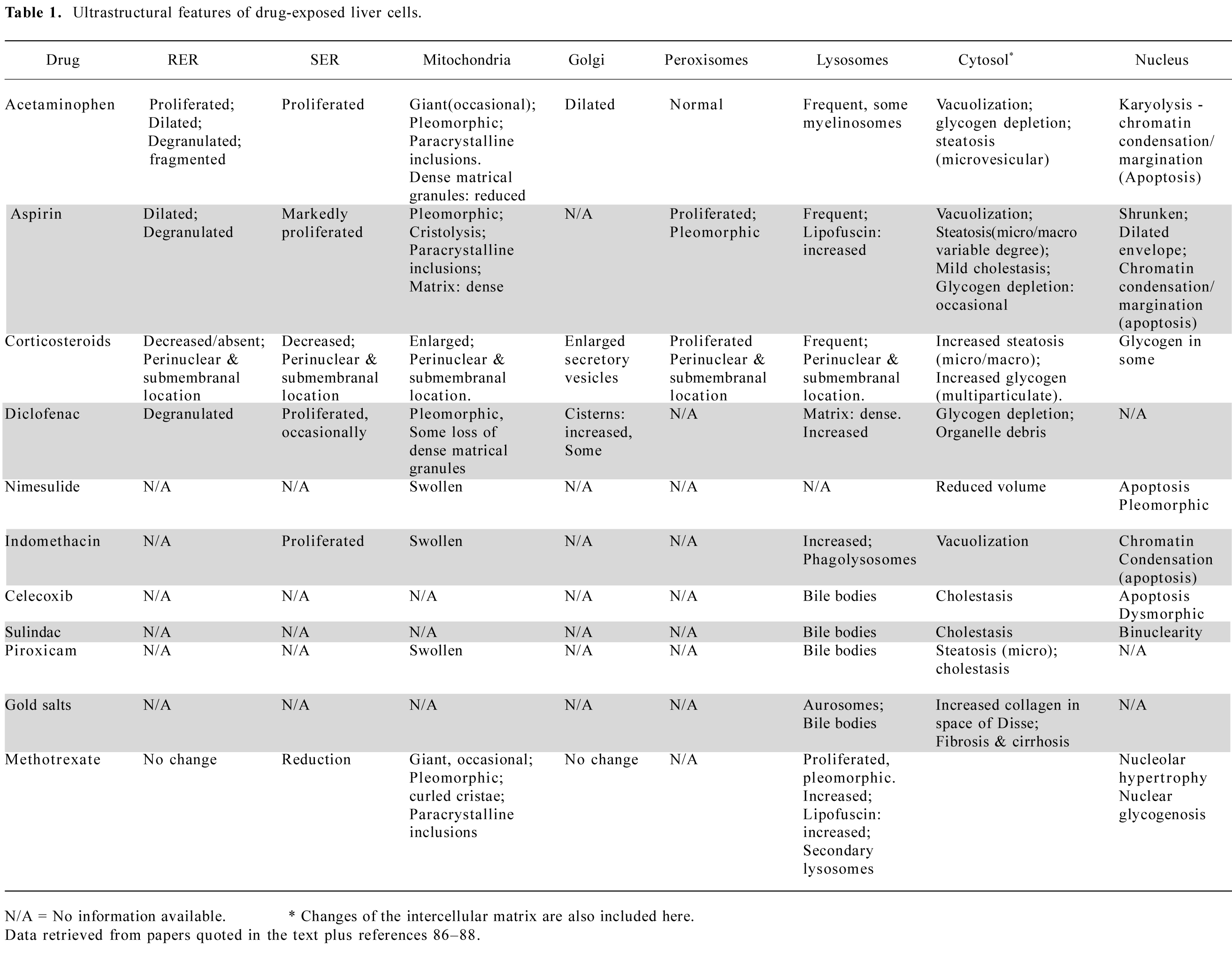
Aspirin/acetylsalicylic acid overdose Since 1956, a number of reports have indicated the elevation of serum transaminases (ALT, AST) in patients treated with aspirin[26–28]. Elevations were noted especially in patients with a serum salicylate concentration higher than 25–30 mg/100 mL for usually 10 or more days. Most of the subjects had rheumatoid arthritis, rheumatic fever, or systemic lupus erythematosus. Light-microscopic observations made by Seaman et al[29] and Iancu et al[30], show preserved liver architecture, but also widened sinusoids and hepatic cytoplasmic vacuolization, indicating degenerative changes. Ultrastructural changes were documented in a single human case[30]. The patient, an 8-year-old boy with rheumatic fever, had elevated transaminase concentrations (AST 220 IU/mL and ALT 240 IU/mL), and a serum salicylate level of 21 mg/mL. Electron microscopic observation of a liver biopsy specimen revealed hepatocytes with shrunken nuclei, and dilated nuclear envelopes with clumping of chromatin (apoptosis). The rough endoplasmic reticulum was dilated and degranulated. The SER had proliferated, and mitochondria were pleomorphic, had increased electron density in the matrix and widened intracristal spaces (Figure 1). These findings led to the immediate discontinuation of aspirin therapy, followed by return to normal of the transaminase levels.
Aspirin and Reye syndrome Reye syndrome (RS), which consists of acute liver failure with encephalopathy, has a potentially fatal outcome (with approximately 40% of affected children dying). The syndrome was frequently diagnosed during 1970–1980, mainly, but not only, in the USA. Initially, RS was considered to be linked to epidemics of varicella and influenza B as well as aspirin ingestion. Indeed, a large group of affected children had increased aspirin levels in their serum. After the use of aspirin diminished, the frequency of RS decreased dramatically. Presently, the rare cases of RS are related to a wide variety of metabolic diseases presenting with a similar clinical and pathological picture. Among these, disorders of oxidative phosphorylation, urea cycle defects, defects in fatty acid oxidation metabolism, systemic carnitine deficiency and acyl-CoA dehydrogenase deficiency, should be noted[31].
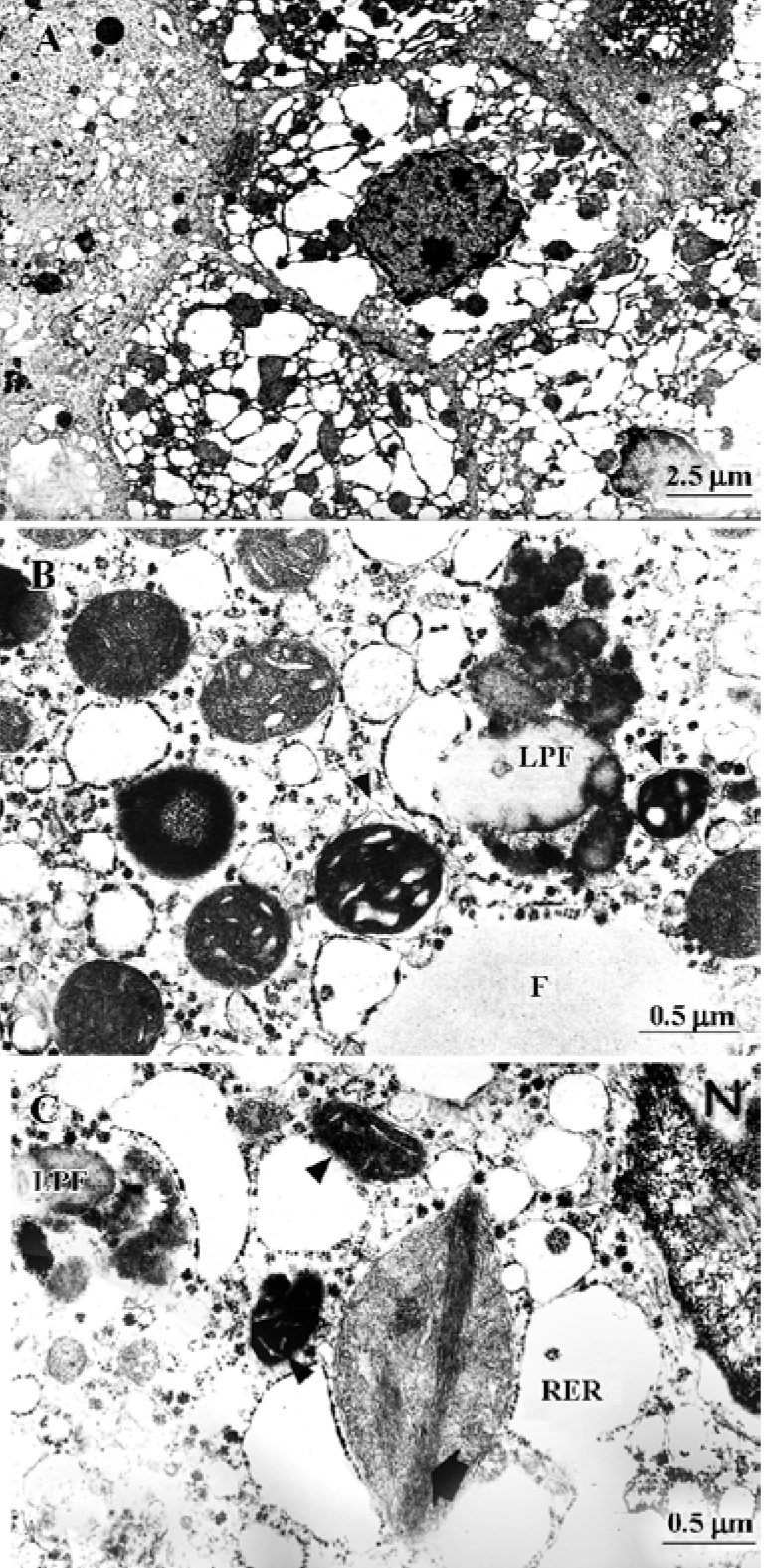
The classical RS, including that related to aspirin ingestion, typically has universal microvesicular hepatocyte steatosis and major ultrastructural changes in the mitochondria. These organelles show marked dilatation, rarefied granular matrix and assume an amoeboid shape. Crystolysis, disappearance of dense matrical bodies and occasional intramitochondrial whorls were also observed. Peroxisomes occasionally appeared with increased frequency, whereas glycogen was depleted. The mitochondrial alterations were less pronounced in the initial stages and were maximal at the height of the disease, when serum ammonia and transaminases were elevated (Figure 2). In parallel, encephalopathy deepened. After this stage, which was reached in 1–2 weeks, some patients recovered spontaneously or with aggressive therapy, and liver function, histological changes and electron microscopic appearance returned to normal[32–34].
Acetaminophen Acetaminophen is widely used as an analgesic and antipyretic with very few adverse effects at therapeutic doses. Hepatotoxicity develops following suicide attempts or after accidental overdose[35]. It has been reported that even borderline high acetaminophen concentrations may induce liver toxicity in infants or in chronic alcoholics[35,36]. The mechanism of liver cell injury produced by acetaminophen overdose remains controversial. Hepatotoxicity is attributed to its transformation into a highly reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), by microsomal enzymes of the P450 family[37]. NAPQI is detoxified by conjugation with glutathione (GSH). Once GSH is depleted, NAPQI covalently binds to proteins, causing alterations in intracellular homeostasis that result in cell necrosis[37–39]. Recently, it was demonstrated that increases in acetaminophen-protein adducts in serum can be used as a predictor of the severity of hepatotoxicity[39]. In addition to liver cell necrosis, acetaminophen can also induce apoptosis[40–45].
Acetaminophen: toxicity in patients There are few studies regarding the ultrastructural changes that occur during acetaminophen overdose. Electron microscopic observations of acetaminophen-induced hepatic toxicity in humans was first described by Dixon et al[46]. These researchers studied the lesions up to 28 d after acetaminophen overdose, and found centrilobular necrosis, hydropic vacuolization, and macrophage infiltration followed by regeneration activity with rapid restoration to normal. In a study of 12 acetaminophen overdoses and 10 other patients with fulminant hepatitis, McCaul et al described differences in the ultrastructural pathology of these groups[47]. Light- and electron microscopic changes were more severe in the fatal non-viral, acetaminophen-induced cases. Acetaminophen overdose elicited prominent changes in hepatocyte nuclei (corresponding to typical changes of apoptosis). Disarrangement of individual hepatocytes due to breakdown in plasma membranes was a frequent finding, as was the presence of amorphous deposits in the endoplasmic reticulum. Depletion of glycogen, mitochondrial swelling and detachment of ribosomes were less prominent in these cases. Comparing his findings with those of Dixon et al. in experimental animals[46], McCaul et al attribute differenences in fine structure to the timing of death in experimental animals versus patients[47]. In our opinion, findings at such an advanced stage of necrosis or necrapoptosis[48] are difficult to interpret because they occurred when necrosis already involved the centrilobular and midzonal cells and, in some cases manifested as massive coagulative necrosis. Biopsies obtained at earlier stages of acetaminophen toxicity are similar to the observations obtained from the liver samples of experimental animals.
Acetaminophen: overdose in experimental animals Attempts to reproduce acetaminophen hepatotoxicity in humans have been made using experimental animals, which were usually given large doses of the drug. Early studies showed initial (3–6 h) depletion of glycogen, loss of ribosomes, cytoplasmic matrix swelling, and mitochondrial abnormalities[49]. After that, as observed mainly in centrilobular cells, there was rapid progression to frank coagulative necrosis. The extensive work of Ray et al addressed the question of the role of hepatocyte apoptosis and necrosis in the process of cellular death[50]. Severe liver injury was produced in ICR mice by administration of 350–500 mg/kg ip acetaminophen. Laboratory and morphological findings were similar to those of humans with acute liver failure following acetaminophen overdose. Ultrastructural examination confirmed the presence of typical apoptotic and necrotic features, similar to the changes found in our later studies with HepG2 and Hep3B cultured hepatoma cells[10,42]. The observation that apoptosis can precede necrosis is of special interest in view of the recent finding that N-acetylcysteine cannot arrest the apoptogenic process[10].
More recently, Ruepp et al detected minor changes at the electron microscopic level as early as 15 min after administration of 500 mg/kg acetaminophen[51]. The changes started in the centrilobular zone and increased in severity and distribution over time. Sixty min later, mitochondrial dilatation was visible as vacuolation by light microscopy. The mitochondria were swollen and fused together. With lower doses (150 mg/kg) the changes were much less severe. Similar findings, also located in the centrilobular zones, were reported by Heinloth et al[52], who exposed rats to 150 mg/kg acetaminophen. No ultrastructural changes were found in controls or rats exposed to 50 mg/kg acetaminophen.
Acetaminophen: in vitro electron-microscopic observations
Isolated rat hepatocytes The effects of acetaminophen on the ultrastructure of isolated rat hepatocytes (IHC) were studied by Fujimura et al[53]. These authors studied suspensions of IHC treated with 5 or 20 mmol/L acetaminophen. The cells had surface blebs containing SER, dilatation of the Golgi apparatus, partial degranulation of the RER and enlargement of the mitochondria. The altered mitochondria had matrix with low electron-density, and with loss of dense matrical granules. The effect of NAPQI, the putative toxic metabolite of acetaminophen, was also investigated: with 500 µmol/L NAPQI, IHC showed surface blebs containing various organelles. Disorderly distributions of cytoplasmic organelles, mild dilatation of RER and SER, and cytoplasmic myeloid bodies were also observed.
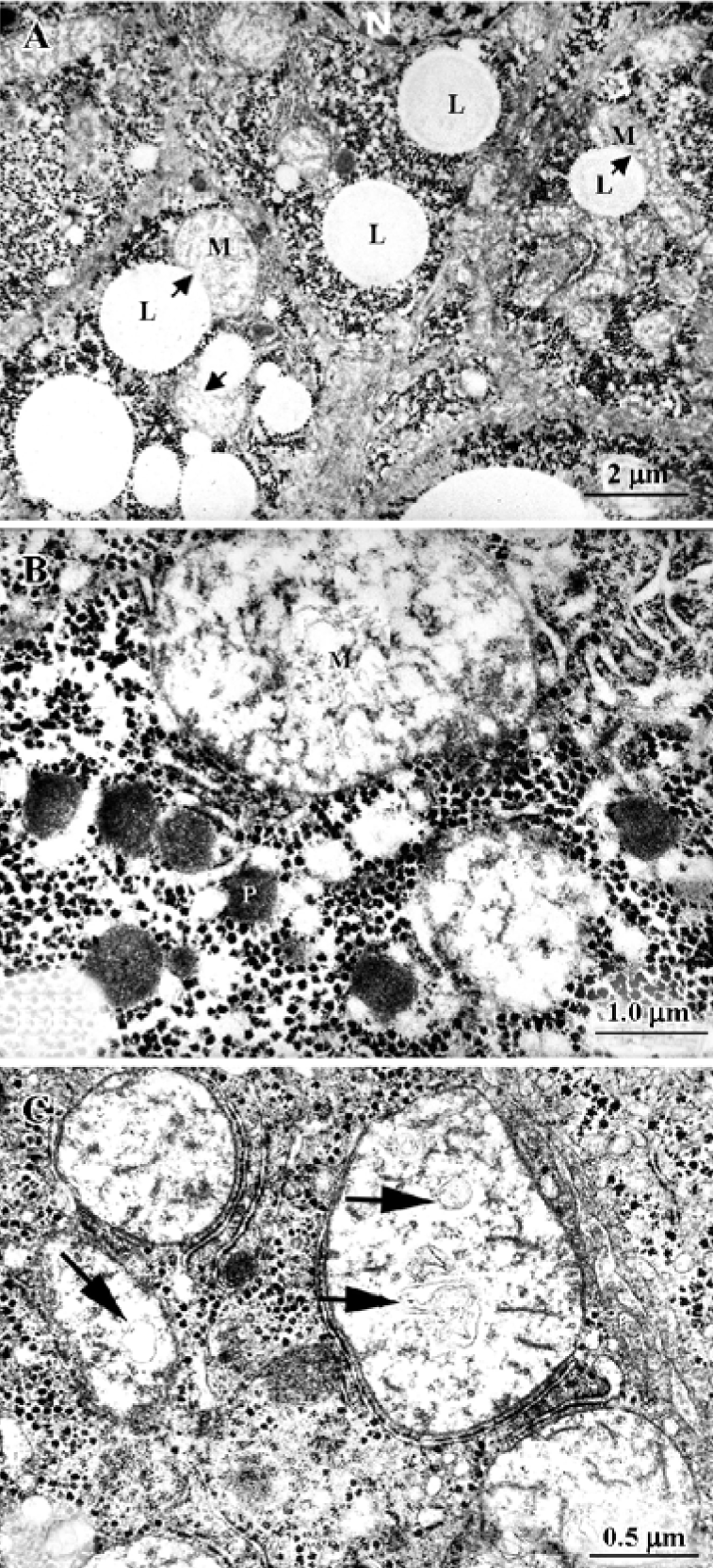
Studies with HepG2 and Hep3B cultured cells We studied the ultrastructure of HepG2 and Hep3B hepatoma-derived cells exposed to acetaminophen for various time periods and at various concentrations[10,42]. There were several types of cells: (1) normal cells, similar to the controls illustrated in Figure 3A and 3B; (2) cells with discrete abnormalities such as reduction in or absence of microvilli, as well as increased frequency of lipid droplets; (3) cells with apoptotic changes (Figure 3C and 3D); (4) typical necrotic cells, with extensive degeneration of the cytoplasm, vacuolization and disruption of the plasma membrane; and (5) so-called “secondary necrotic” cells with morphological elements of both apoptosis and necrosis (Figure 3E).
Variability in the frequency of mitochondrial dense granules in HepG2 cells exposed or not to acetaminophen was also studied by Manov et al[10]. After acetaminophen treatment, the percentage of mitochondrial dense granule-positive mitochondria decreased significantly. The disappearance of mitochondrial dense granules has been included among the reversible changes in acute hepatotoxicity[4], but their absence has also been observed in other conditions[13].
Morphological changes associated with apoptosis such as cell shrinkage, chromatin condensation and margination, and apoptotic bodies were also observed in the liver after high doses of acetaminophen[40,54]. Quantitative analysis performed by Ray et al on mouse liver indicated that 40% of cells died by apoptosis 24 h after acetaminophen administration[50,55]. In contrast, Gujral et al showed that necrosis, but not apoptosis, is the predominant mechanism of cell death after acetaminophen overdose in vivo[54]. Because apoptotic cells in vivo are rapidly removed from the tissue by phago-cytes, this data may be an underestimate. According to our in vitro findings, both apoptosis and necrosis are observed in HepG2 and Hep3B cells after acetaminophen treatment[10,42]. To differentiate apoptotic from necrotic cells, we used ultrastructural morphometry and cell staining with annexin V and propidium iodide. We found that after 24 h exposure to 10 mmol/L acetaminophen, HepG2 cells died predominantly by apoptosis, whereas necrosis was prominent after 48 h. In this late stage of toxicity the apoptotic process may switch to secondary necrosis and apoptotic cells may become indistinguishable from necrotic ones[56]. Additionally, apoptosis in vitro in the absence of phagocytes may normally degenerate to secondary necrosis[57].
In our in vitro investigation we found that N-acetyl-cysteine, a known antioxidant used in acute acetaminophen poisoning, does not prevent acetaminophen-induced apoptosis and necrosis[10,42]. Exposure of Hep3B cells to acetaminophen and N-acetylcysteine for 48 h resulted in an increase in apoptotic cells in comparison with cells treated with acetaminophen alone. In HepG2 cells 5 mmol/L of N-acetylcysteine markedly increased the degree of necrosis and secondary necrosis in comparison with cells treated with acetaminophen only. N-Acetylcysteine alone did not induce apoptotic or necrotic changes in HepG2 or Hep3B cells.
An interesting combination of hepatotoxic factors, namely acetaminophen and ethanol, was studied by Neuman[44]. Electron microscopy of normal human primary hepatocytes treated with 40 mmol/L ethanol plus 10 mmol/L acetaminophen for 24 h demonstrated that the treated hepatocytes had a common specific attribute: their mitochondria were enlarged and appeared swollen or elongated, with disrupted cristae, and lacked normal organization. The combination of 10 mmol/L acetaminophen and 40 mmol/L ethanol was synergistic in producing mitochondrial damage after 24 h. Giant mitochondria, 3 µm in length, were observed within cells exposed to this treatment. The endoplasmic reticulum underwent enlargement and vesiculation, and lipid accumulation was also seen.
The efflux pump P-glycoprotein (P-gp) has been shown to have an important role in intracellular drug concentration. To evaluate the contribution of the P-gp transporter to the course of acetaminophen-induced toxicity, we compared toxicity to cells caused by acetaminophen alone, and acetaminophen plus verapamil, a well known inhibitor of P-gp activity. If P-gp is involved in acetaminophen transport, toxicity would be increased by inhibition of the P-gp pump activity. Among other parameters, we are presently studying the ultrastructural changes caused by these agents. Preliminary observations show that after exposure of HepG2 and Hep3B cells to verapamil (100 µmol/L for 24 h) numerous single-membrane-limited bodies with variable content (autophagolysosomes) were seen, whereas other subcellular features remained apparently intact. Treatment with acetaminophen plus verapamil increased the frequency of necrotic cells. In a “prenecrotic” stage, cells displayed extensive SER dilatation/proliferation (Figure 3F) in parallel with other features of damage (eg autophagolysosomes, reduction of surface microvilli, and increased vacuolization). On the basis of this series of experiments we suspect that P-gp is involved in acetaminophen transport and may play a significant role in acetaminophen-induced toxicity.
Nonsteroidal anti-inflammatory drugs The heterogeneity of this group notwithstanding, NSAIDs have common therapeutic effects and similar side-effects. Individual drugs only very rarely have noxious effects, but their extremely widespread use enhances the hazard. Despite their common toxic effects, individual features have been noticed among various NSAIDs with respect to the liver pathology induced. For example the following pathologies are induced by the drugs indicated: granulomatous hepatitis (phenylbutazone), hepatonecrotic lesions (phenylbutazone, sulindac, diclofenac, pirprofen, piroxicam), and cholestatic hepatitis (sulindac, benoxprofen, ibuprofen, phenylbutazone, piroxicam).
Diclofenac The scarce side effects of diclofenac are significant because of its world-wide use by many patients. Therapeutic use of diclofenac is associated with rare but sometimes fatal hepatotoxicity characterized by delayed onset of symptoms and lack of a clear dose–response relationship. The toxicity has consequently been categorized as metabolic idiosyncrasy. In fact, the acyl glucuronide of the drug has been demonstrated to be reactive and capable of covalent modification of cellular proteins, binding covalently to liver proteins in rats depending on the activity of multidrug resistance protein 2, a hepatic canalicular transporter[58]. From a toxicodynamic point of view, both oxidative stress (caused by putative diclofenac cation radicals or nitroxide and quinone imine-related redox cycling) and mitochondrial injury (protonophoretic activity and opening of the permeability transition pore) alone or in combination, have been implicated in diclofenac toxicity. In some cases, immune-mediated liver injury is involved, as inferred from inadvertent rechallenge data and from a number of experiments demonstrating T cell sensitization. Caspases 8 and 9 are apparently active caspases in diclofenac-induced apoptosis. In addition, an early dose-dependent increase of Bcl-X L expression parallel to the generation of reactive oxygen species in the mitochondria was found. In conclusion, the mitochondrial pathway is very likely the only pathway involved in diclofenac-induced apoptosis, which is related to CYP-mediated metabolism of diclofenac, with the highest apoptotic effect produced by the metabolite 5OH-diclofenac. To date, cumulative damage to mitochondrial targets seems a plausible putative mechanism to explain the delayed onset of liver failure, perhaps even superimposed on an underlying silent mitochondrial abnormality[58]. Although Gomez-Lechon et al have demonstrated that diclofenac induces apoptosis by alteration of mitochondrial function and generation of reactive oxygen species (ROS)[59], liver injury could not be reproduced in current animal models. Nevertheless, it is noteworthy that ultrastructural damage was found in the liver of rainbow trout exposed to various concentrations of diclofenac, thus illustrating differences in reactivity between mammals and other vertebrates[60].
Nimesulide Nimesulide is a selective cyclo-oxygenase-2 (COX-2) inhibitor that has been recently linked with rare but serious and unpredictable adverse reactions in the liver, such as fulminant hepatic failure. It is thought that the hepatotoxicity induced by nimesulide is of the idiosyncratic type. In addition, the weakly acidic sulfonanilide drug undergoes bioreductive metabolism of the nitroarene group to form reactive intermediates that have been implicated in oxidative stress, covalent binding, and mitochondrial injury[61]. Tian et al investigated the effect of nimesulide on the proliferation and apoptosis of SMMC-7721 human hepatoma cells[62]. Under the electron microscope, these cells exhibited characteristics of apoptosis, including plasma membrane blebbing, cytoplasmic condensation, pyknotic nuclei, condensed chromatin and apoptotic bodies. Compared with control groups, groups treated with 300 µmol/L and 400 µmol/L nimesulide had many more cells with apoptotic characteristics. This observation is of interest because of the putative antitumor effect of NSAIDs.
Indomethacin The incidence of liver injury associated with indomethacin appears to be low, although exact figures are not available. The reactions usually develop within the first few months of treatment, sometimes in association with manifestations of hypersensitivity, and are fatal in rare instances. The histological findings range from an acute viral-like hepatitis to confluent or multilobular necrosis; chronic active hepatitis with cholestasis and fatty changes are also occasionally observed. Sorensen and Acosta reported that cells exposed to high concentrations of indomethacin were severely damaged, as evidenced by marked cellular necrosis, nuclear pleomorphism, margination of chromatin, swollen mitochondria, reductions in the number of microvilli, smooth endoplasmic reticulum proliferation, and cytoplasmic vacuolation[63].
Celecoxib Celecoxib, a specific COX-2 enzyme inhibitor, has been widely used to alleviate pain and inflammation in osteoarthritis and rheumatoid arthritis. It has minimal gastrointestinal, platelet, and renal side effects, but has been associated with acute hepatocellular and cholestatic injury[64]. Results of an in vitro study indicate that celecoxib may inhibit proliferation and induce apoptosis in human cholan-giocarcinoma cells through its effect on COX-dependent mechanisms and the PGE2 pathway. Celecoxib as a chemo-preventive and chemotherapeutic agent may be primarily effective for COX-2-expressing cholangiocarcinoma, but may also be effective for other tumors as well[65].
Ibuprofen Patients with hepatitis C may show increased serum transaminase concentrations when treated with ibuprofen. One morphological feature that deserves to be mentioned here is the “vanishing bile duct syndrome”, which has been reported in children with chronic rejection after transplantation[21,66], and also in adults, and has been associated with more than 30 drugs, including ibuprofen[67]. More severe reactions, such as subfulminant hepatitis requiring liver transplantation following ibuprofen overdose, although very rare, have also been described[68].
Naproxen As with other NSAIDs, borderline abnormalities in one or more liver tests may occur in up to 15% of patients treated with naproxen. The ALT test is probably the most sensitive indicator of liver dysfunction. Meaningful (3 times the normal upper limit) elevations of ALT or AST occurred in controlled clinical trials in less than 1% of patients. Severe hepatic reactions, including jaundice and cases of fatal hepatitis, have been reported with naproxen as with other NSAIDs[69]. Although such reactions are rare, if abnormal liver tests persist or worsen, and if clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (eg eosinophilia or rash), naproxen treatment should be discontinued[70].
Sulindac Sulindac has been associated with various toxic side-effects, including neurological, hematological, renal and liver damage. From a histopathological perspective, cholestatic and/or hepatocellular damage are the morphologic expressions of toxicity. Tarazi et al analyzed 91 cases of sulindac-associated hepatic injury, among which there were four fatal outcomes[71]. The morphological features of sulindac hepatotoxicity were reported by Wood and co-workers[72], who described a cholestatic hepatitis with marked anisonucleosis, cytoplasmic invaginations into the nucleus and binuclearity of hepatocytes.
Piroxicam Isolated severe cholestatic[73] and fatal[74] cases have been reported in various age groups in response to piroxicam treatment. Liver morphology in these cases indicated acute hepatitis with cholestasis and confluent necrosis. Microvesicular steatosis has also been reported.
Disease-modifying antirheumatic drugs
The disease-modifying antirheumatic drug (DMARD) group comprises both chemical and biological (immunosupp-ressive) compounds. In recent years, biological compounds have been used with increased frequency, mainly in rheumatoid arthritis. Infliximab, anakinra, etanercept and leflunomide are the major representatives of this group. For leflunomide, hepatotoxicity, defined as a 3-fold increase in ALT above the normal upper limit, has been found in up to 10% of patients, but cases of acute liver failure have been rare. The pathological findings included a variety of abnormalities, including centrilobular necrosis with portal or periportal inflamma-tion, steatosis, focal piecemeal necrosis, and periportal fibrosis. Too few ultrastructural observations have been reported to enable a meaningful description. However, among DMARDs, gold compounds and methotrexate (MTX) deserve further discussion because of their longer use and better-known hepatotoxicity[75].
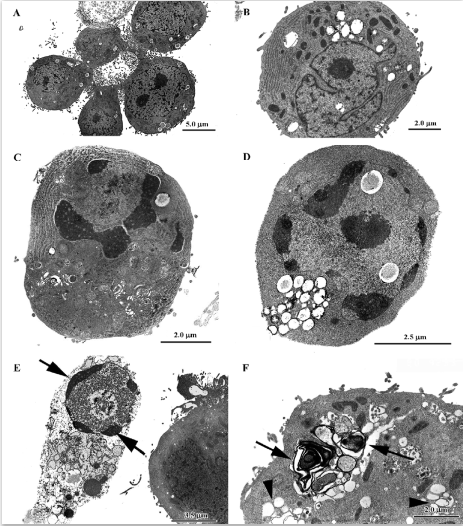
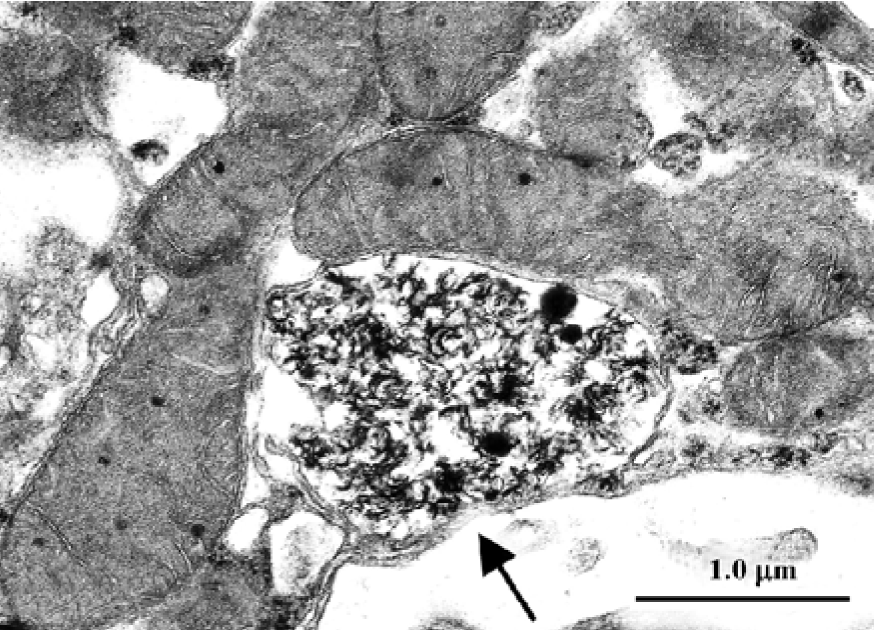
Gold hepatotoxicity Among DMARDs, gold continues to have a place, although it is now seldom used. Although the more frequent complications of crysotherapy are gold nephritis and dermatitis, occasional cases of severe, even lethal hepatotoxicity are still reported[76]. When transaminase concentrations increase, liver biopsy becomes compul-sory. In gold nephropathy, electron microscopy reveals typical lysosomes, which are termed aurosomes[13] (Figure 4; TC Iancu and O Ben-Itzhak, unpublished micrograph), but gold deposits can be found in various other organs, including the liver, where they induce fibrosis and cirrhosis. Histopatholo-gically, inflammation and severe loss of parenchyma can be seen, while macrophages contain dark granules that are shown by electron microscopy to be typical aurosomes[77].
Methotrexate Methotrexate is presently used as the initial DMARD, especially for patients whose rheumatoid arthritis is more active. It is frequently effective and has low toxicity. During recent years, various regimens of MTX therapy have been used, and its side-effects have become better known[78]. The macrovesicular steatosis, fibrosis and cirrhosis that were noted earlier are now rare because MTX is not given in daily but weekly doses. However, hepatitis with bridging fibrosis[79] and methotrexate-induced cirrho-sis[80] were still reported until 1990. Phillips et al described several liver ultrastructural changes in MTX-treated patients (Table 1)[1].
Earlier studies (in 1988) showed that patients with rheumatoid arthritis treated with MTX had an increased frequency of abnormal, pleomorphic lysosomes and increased collagen in the space of Disse[81]. A year later, Kremer and Kaye showed that these features were present also in untreated patients[82]. Their observations were reinforced in 1995, when they reported that there was no significant increase in collagen deposition after prolonged MTX therapy[83]. Ros et al carried out a light and electron microscopic analysis of 42 liver biopsies from patients with rheumatoid arthritis treated for 4 years with MTX (weekly doses of 7.5–15 mg)[84]. The availability of pre-MTX biopsies enabled a valid comparison with post-MTX findings. Electron microscopy appeared to be more sensitive for identifying increases in collagen fibers in the space of Disse. By using light microscopy, the authors concluded that 14% of rheumatoid arthritis patients had mild perisinusoidal fibrosis before MTX therapy, whereas electron microscopy revealed increased collagen presence in the space of Disse in 50%. After 4 years of MTX therapy, no significant increase in fibrosis was found. The authors concluded that MTX therapy is safe, regardless of the mild fibrotic changes found in the initial biopsies. Recently, Maurice et al also stressed the minimal toxicity of low-dosage, long-term MTX therapy[85]. These authors monitored patients on MTX with normal serum assays of the amino terminal peptide of type III procollagen, and found no hepatic fibrosis on biopsy. Maurice et al concluded that liver biopsy could be avoided in 45% of cases.
Conclusions
The study of ultrastructural changes in liver cells following exposure to xenobiotics or excessive doses of medication is helpful for:
1) Detection of diagnostic features, particularly in complex syndromes (specific changes are scarce; various agents mostly produce similar features of damage).
2) Assessment of the degree of damage, from mild, reversible changes to severe, irreversible (apoptosis, necrapoptosis, necrosis) changes, which are usually related to duration of exposure and concentration.
3) Identification of target-specificity: there are marked differences in the subcellular changes produced by the same agent in different cell types, as seen in cultures of primary cells (eg hepatocytes), tumoral cells (eg HepG2, Hep3B) and biopsy samples (from humans or experimental animals).
The information provided by ultrastructural study has contributed to the unraveling of the mechanisms of hepatotoxicity. Electron microscopy continues to be an essential tool for the study of drug-induced liver diseases.
References
- Phillips JM, Poucell S, Patterson J, Valencia P. The liver. New York: Raven Press; 1979.
- Rappaport AM. Physioanatomic considerations. In: Schiff L, Schiff ER editors. Disease of the liver. 5th edition. Philadelphia: J B Lippincott; 1982; 1-57.
- Trump BF, Berezesky IK, Osornio-Vargas AR. Cell death and the disease process. In: Bowen ID, Lockshin RA, editors. Cell death in biology and pathology. London: Chapman & Hall 1981; 209–42.
- Desmet VJ, Vos DE. Structural analysis of acute liver injury. In: Keppler D, Popper H, Bianchi L, Reutter W, editors. Mechanisms of hepatocyte injury and death. Lancaster: MTP Press Limited 1984; 11–30.
- Cheville NF. Ultrastructural pathology: An introduction to interpretation. Ames, IA: Iowa State University Press; 1994.
- Pompella A, Comporti M. Imaging of oxidative stress at subcellular levels by confocal laser scanning microscopy after fluorescent derivativization of cellular carbonyls. Am J Pathol 1993;142:1353-7.
- Slot JW, Geuze HJ, Freeman BA, Crapo JD. Intracellular localization of the copper-zinc and manganese superoxide dismutases in rat liver parenchymal cells. Lab Invest 1986;55:363-71.
- Rieder IH, Teutsch HF, Sasse D. NADP-dependent dehydrogenases in rat liver parenchyma. Histochemistry 1978;56:283-98.
- Lee SH, Torack RM. A biochemical and histochemical study of glutamic acid oxalacetic transaminase activity of rat hepatic mitochondria fixed in situ and in vitro. J Cell Biol 1968;39:725-32.
- Manov I, Hirsh M, Iancu TC. N-Acetylcysteine does not protect HepG2 cells against acetaminophen-induced apoptosis. Basic Clin Pharmacol Toxicol 2004;94:213-25.
- Trump BF, Goldblatt PJ, Stowell RE. Studies on necrosis of mouse liver in vitro: ultrastructural alterations in the mitochondria of hepatic parenchymal cells. Lab Invest 1965;14:2000-28.
- Greene WB, Balentine JD, Hennigar GR. Selective mitochondrial degeneration in renal tubules following hyperbaric oxygen exposure. Am J Pathol 1979;96:737-52.
- Ghadially FN. Ultrastructural pathology of the cell and matrix. 4 ed. Boston: Butterworth-Heinemann; 1997.
- Russo MA, van Rossum GD. The basis for the cellular damage induced by ethacrynic acid in liver slices in vitro. Lab Invest 1986;54:695-707.
- Keller GA, Scallen TJ, Clarke D, Maher PA, Krisans SK, Singer SJ. Subcellular localization of sterol carrier protein-2 in rat hepatocytes: Its primary localization in peroxisomes. J Cell Biol 1989;108:1353-61.
- Nicholls-Grzemski FA, Burcham PC, Calder IC, Priestly BG. Pretreatment with peroxisome proliferators protect mice against some but not all hepatotoxins. In: Reddy JK, Suga T, Mannaerts GP, Lazarow PB, Subramani S, editors. Peroxisomes: Biology and role in toxicology and disease. New York: The New York Academy of Sciences; 1996. p 742–4.
- Peterlik M, Kerjaschki D. Interference of tris(hydroxymethyl)aminomethane with structure and function of Golgi membranes. Lab Invest 1979;40:313-6.
- Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther 2001;92:57-70.
- Levanon D, Manov I, Iancu TC. Qualitative and quantitative analysis of the effects of acetaminophen and N-acetylcysteine on the surface morphology of Hep3B hepatoma cells in vitro. Ultrastruct Pathol 2004;28:3-14.
- Schneider P, Korolenko TA, Busch U. A review of drug-induced lysosomal disorders of the liver in man and laboratory animals. Microsc Res Tech 1997;36:253-75.
- Zimmerman HJ, Ishak KG. Hepatic injury due to drugs and toxins. In: MacSween RNM, Anthony PP, Scheuer PJ, Burt AD, Portmann BC, editors. Pathology of the liver. Edinburgh: Churchill Livingstone; 1994; p 563–633.
- Lewis HJ. Drug-induced liver disease. Med Clin North Am 2000;84:1275-311.
- Alpers DH, Sabesin SM. Fatty liver: Biochemical and clinical aspects. In: Schiff L, Schiff ER, editors. Diseases of the liver. Philadelphia: JB Lippincott; 1982. p 813–45.
- Iancu TC, Shiloh H, Dembo L. Hepatomegaly following short-term high-dose steroid therapy. J Pediatr Gastroenterol Nutr 1986;5:41-6.
- Baxter JD, Forsham PH. Tissue effects of glucocorticoids. Am J Med 1972;53:573-89.
- Tanikawa K. Ultrastructural aspects of the liver and its disorders. 2nd ed. Tokyo: Igaku-Shoin; 1979.
- Manso C, Nydick I, Taranta A. Effects of aspirin administration on serum glutamic oxaloacetic and glutamic pyruvic transaminases in children. Proc Soc Exp Biol Med 1956;93:84-8.
- Iancu T, Elian E. Aspirin induced abnormalities of liver function Am J Dis Child 1974;128:116-7. [letter].
- Seaman WE, Ishak KG, Plotz PH. Aspirin induced hepatotoxicity in patients with systemic lupus erythematosus. Ann Intern Med 1974;80:1-8.
- Iancu T, Elian E. Ultrastructural changes in aspirin hepato-toxicity. Am J Clin Pathol 1976;66:570-5.
- Chakrapani A, Green A. Metabolic liver diseases in the infant and older child. In: Kelly DA, editor. Disease of the liver and biliary system. Melbourne: Blackwell Publishing; 2004. p 211–42.
- Reye RDK, Morgan G, Baral J. Encephalopathy and fatty degeneration of the viscera: a disease entity in childhood. Lancet 1963;2:749-52.
- Partin JC, Schubert WK, Partin JS. Mitochondrial ultrastructure in Reye’s syndrome (encephalopathy and fatty degeneration of the viscera). N Engl J Med 1971;285:1339-43.
- Iancu TC, Manson WH, Neustein HB. Ultrastructural abnormalities of liver cells in Reye’s syndrome. Hum Pathol 1977;4:421-31.
- Zimmerman HJ, Maddrey WC. Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: analysis of instances of therapeutic misadventure. Hepatology 1995;22:767-73.
- Kearns GL, Leeder JS, Wasserman GS. Acetaminophen intoxication during treatment: what you don’t know can hurt you. Clin Pediatr (Phila) 2000;39:133-44.
- Nelson SD. Mechanisms of the formation and disposition of reactive metabolites that can cause acute liver injury. Drug Metab Rev 1995;27:147-77.
- Pumford NR, Halmes NC, Hinson JA. Covalent binding of xenobiotics to specific proteins in the liver. Drug Metab Rev 1997;29:39-57.
- Muldrew KL, James LP, Coop L, McCullough SS, Hendrickson HP, Hinson JA, et al. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos 2002;30:446-51.
- Ray SD, Jena N. A hepatotoxic dose of acetaminophen modulates of BCL-2, BCL-X (L), and BCL-X(S) during apoptotic and necrotic death of mouse liver cells in vivo. Arch Toxicol 2000;73:594-606.
- Ferret PJ, Hammoud R, Tulliez M, Tran A, Trebeden H, Jaffray P, et al. Detoxification of reactive oxygen species by a nonpe-ptidyl mimic of superoxide dismutase cures acetaminophen-induced acute liver failure in the mouse. Hepatology 2001;33:1173-80.
- Manov I, Hirsh M, Iancu TC. Acetaminophen hepatotoxicity and mechanisms of its protection by N-acetylcysteine: a study of Hep3B cells. Exp Toxicol Pathol 2002;53:489-500.
- xxxxx.
- Neuman MG. Synergetic signaling for apoptosis in vitro by ethanol and acetaminophen. Alcohol 2002;27:89-98.
- El-Hassan H, Anwar K, Macanas-Pirard P, Crabtree M, Chow SC, Johnson VL, et al. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, Bax, Bid, and caspases. Toxicol Appl Pharmacol 2003;191:118-29.
- Dixon MF, Nimmo J, Prescott LF. Experimental paracetamol-induced hepatic necrosis: a histopathological study. J Pathol 1971;103:225-9.
- McCaul T, Fagan E, Tovey G, Portmann B, Williams R, Zuckerman AJ. Fulminant Hepatitis: An ultrastructural study. J Hepatol 1986;368:276-290.
- Lemasters JJ. Mechanisms of hepatic toxicity V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Gastrointest Liver Physiol 1999;39:G1-G6.
- Dixon M, Dixon B, Aparicio S, Loney D. Experimental paracetamol-induced hepatic necrosis: A light- and electron-microscope, and histochemical study. J Pathol 1975;116:17-29.
- Ray S, Mumaw V, Raje R, Fariss M. Protection of acetaminophen-induced hepatocellular apoptosis and necrosis by cholesteryl hemisuccinate pretreatment. J Pharmacol Exp Ther 1996;279:1470-83.
- Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci 2002;65:135-50.
- Heinloth AN, Irwin RD, Boorman GA, Nettesheim P, Fannin RD, Sieber SO, et al. Gene expression profiling of rat livers reveals indicators of potential adverse effects. Toxicol Sci 2004;80:193-202.
- Fujimura H, Kawasaki N, Tanimoto T, Sasaki H, Suzuki T. Effects of acetaminophen on the ultrastructure of isolated rat hepatocytes. Exp Toxicol Pathol 1995;47:345-51.
- Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci 2002;67:322-8.
- Ray SD, Kumar MA, Bagchi D. A novel proanthocyanidin IH636 grape seed extract increases in vitro Bcl-XL expression and prevents acetaminophen-induced programmed and unprogrammed cell death in mouse liver. Arch Biochem Biophys 1999;369:42-58.
- Levin S, Bucci TJ, Cohen SM, Fix AS, Hardisty JF, LeGrand EK, et al. The nomenclature of cell death: recommendations of an ad hoc Committee of the Society of Toxicologic Pathologists. Toxicol Pathol 1999;27:484-90.
- Gomez-Lechon MJ, O’Connor E, Castell JV, Jover R. Sensitive markers used to identify compounds that trigger apoptosis in cultured hepatocytes. Toxicol Sci 2002;65:299-308.
- Tang W. The metabolism of diclofenac-enzymology and toxicology perspectives. Curr Drug Metab 2003;4:319-29.
- Gomez-Lechon MJ, Ponsoda X, O’Connor E, Donato T, Castell JV, Jover R. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem Pharmacol 2003;66:2155-67.
- Triebskorn R, Casper H, Heyd H, Eikemper R, Köhler HR, Schwaiger J. Toxic effects of the non-steroidal anti-inflammatory drug diclofenac Part II. Cytological effects in liver, kidney, gills and intestine of rainbow trout (Oncorhynchus mykiss). Aquat Toxicol 2004;68:151-66.
- Boelsterli UA. Mechanisms of NSAID-induced hepatotoxicity: focus on nimesulide. Drug Saf 2002;25:633-48.
- Tian G, Yu JP, Luo HS, Yu BP, Yue H, Li JY, et al. Effect of Nimesulide on proliferation and apoptosis of human hepatoma SMMC-7721 cells. World J Gastroenterol 2002;8:483-7.
- Sorensen EM, Acosta D. Relative toxicities of several nonsteroidal anti-inflammatory compounds in primary cultures of rat hepatocytes. J Toxicol Environ Health 1985;16:425-40.
- Nachimuthu S, Volfinzon L, Gopal L. Acute hepatocellular and cholestatic injury in a patient taking celecoxib. Postgrad Med J 2001;77:548-50.
- Koki AT, Masferrer JL. Celecoxib: A specific COX-2 inhibitor with anticancer properties. Cancer Control 2002;9 Suppl 2:28-35.
- Kelly DA, Mayer D. Liver transplantation. In: Kelly DA, editor. Diseases of the liver and biliary system. Melbourne: Blackwell Publishing; 2004. p 378–401.
- Mayoral W, Lewis JH, Zimmerman H. Drug-induced liver disease. Curr Opin Gastroenterol 1999;15:208-16.
- Laurent S, Rahier J, Geubel AP, Lerut J, Horsmans Y. Subfulminant hepatitis requiring liver transplantation following ibuprofen overdose. Liver 2000;20:93-4.
- Giarelli L, Falconieri G, Delendi M. Fulminant hepatitis following naproxen administration. Hum Pathol 1986;17:1079. Erratum in: Hum Pathol 1987; 18: 205.
- Mehta N, Osick L. Drug-induced hepatotoxicity [page on Internet]. eMedicine updated 2 November 2005; Available from: http://www.emedicine.com/med/topic3718.htm
- Tarazi EM, Harter JG, Zimmerman HJ, Ishak KG, Eaton RA. Sulindac-associated hepatic injury: analysis of 91 cases reported to the Food and Drug Administration. Gastroenterology 1993;104:569-74.
- Wood LJ, Mundo F, Searle J, Powell LW. Sulindac hepatotoxicity: effects of acute and chronic exposure. Aust NZ J Med 1985;15:397-401.
- Poniachik J, Guerrero J, Calderon P, Smok G, Morales A, Munoz G, et al. Cholestatic hepatitis associated with piroxicam use. Case report. Rev Med Chil 1998;126:548-52.
- Rabinovitz M, Van Thiel DH. Hepatotoxicity of nonsteroidal anti-inflammatory drugs. Am J Gastroenterol 1992;87:1696-704.
- Felson DT, Anderson JA, Meenan RF. The comparative efficacy and toxicity of second line drugs in rheumatoid arthritis. Results of 2 meta-analyses. Arthritis Rheum 1990;10:1449-61.
- Watkins PB, Schade R, Mills AS, Carithers RL Jr, Van Thiel DH. Fatal hepatic necrosis associated with parenteral gold therapy. Dig Dis Sci 1988;33:1025-9.
- Fleischner GM, Morecki R, Hanaichi T, Hayashi H, Quintana N, Sternlieb I. Light- and electron-microscopical study of a case of gold salt-induced hepatotoxicity. Hepatology 1991;14:422-5.
- van Ede AE, Laan RF, Blom HJ, De Abreu RA, van de Putte LB. Methotrexate in rheumatoid arthritis: an update with focus on mechanisms involved in toxicity. Semin Arthritis Rheum 1998;27:277-92.
- Kujala GA, Shamma’a JM, Chang WL, Brick JE. Hepatitis with bridging fibrosis and reversible hepatic insufficiency in a woman with rheumatoid arthritis taking methotrexate. Arthritis Rheum 1990;33:1037-41.
- Gilbert SC, Klintmalm G, Menter A, Silverman A. Methotrexate-induced cirrhosis requiring liver transplantation in three patients with psoriasis. A word of caution in light of the expanding use of this ‘steroid-sparing’ agent. Arch Intern Med 1990;150:889-91.
- Bjorkman DJ, Hammond EH, Lee RG, Clegg DO, Tolman KG. Hepatic ultrastructure after methotrexate therapy for rheumatoid arthritis. Arthritis Rheum 1988;31:1465-72.
- Kremer JM, Kaye GI. Electron microscopic analysis of sequential liver biopsy samples from patients with rheumatoid arthritis. Correlation with light microscopic findings. Arthritis Rheum 1989;32:1202-13.
- Kremer JM, Kaye GI, Kaye NW, Ishak KG, Axiotis CA. Light and electron microscopic analysis of sequential liver biopsy samples from rheumatoid arthritis patients receiving long-term methotrexate therapy. Follow-up over long treatment intervals and correlation with clinical and laboratory variables. Arthritis Rheum 1995;38:1194-203.
- Ros S, Juanola X, Condom E, Canas C, Riera J, Guardiola J, et al. Light and electron microscopic analysis of liver biopsy samples from rheumatoid arthritis patients receiving long-term methotrexate therapy. Scand J Rheumatol 2002;31:330-6.
- Maurice PDL, Maddox AJ, Green CA, Tatnall F, Shofield JK, Stott DJ. Monitoring patients on methotrexate: hepatic fibrosis not seen in patients with normal serum assays of aminoterminal peptide of type III procollagen. Br J Dermatol 2005;152:451-8.
- Sternlieb I. Electron microscopy of mitochondria and peroxisomes of human hepatocytes. Prog Liver Dis 1979;6:81-104.
- Salgueiro-Pagadigorria CL, Kelmer-Bracht AM, Bracht A, Ishii-Iwamoto EL. Effects of the nonsteroidal anti-inflammatory drug piroxicam on rat liver mitochondria. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 1996;113:85-91.
- Caparroz-Assef SM, Bersani-Amado CA, do Nascimento EA, Kelmer-Bracht AM, Ishii-Iwamoto EL. Effects of the nonsteroidal anti-inflammatory drug nimesulide on energy metabolism in livers from adjuvant-induced arthritic rats. Res Commun Mol Pathol Pharmacol 1998;99:93-116.