
Neuroprotective effects of stearic acid against toxicity of oxygen/glucose deprivation or glutamate on rat cortical or hippocampal slices1
Introduction
Brain is rich in diverse fatty acids (saturated, monoun-saturated and polyunsaturated), with the complex lipids that are present having chain lengths ranging from less than 16 to more than 24 carbons. Previously, exploration of long-chain fatty acids has placed emphasis on investigating their fate after they are metabolized by cells. For example, intracellular fatty acids are processed into phospholipids, possibly affecting certain membrane functions, or are used to supply energy after decomposition by mitochondria[1]. The body’s preferred energy source is glucose, which comes from carbohydrates, but it can also use fatty acids and amino acids. However, the brain relies almost exclusively on glucose for oxidative ATP production.
Epidemiological, clinical and biochemical studies have shown that different types of dietary fatty acids can modify the risks of stroke[2,3], but the molecular and cellular mechanisms by which dietary fatty acids exert such effects are still not well understood. Polyunsaturated fatty acids (PUFA), specifically the n-3 series, have been implicated in the prevention of various human diseases, including obesity, diabetes, coronary artery disease and stroke, and inflammatory and neurological diseases[4]. Only a few articles have reported the effects of saturated fatty acids on human health. Saturated fats are usually regarded as unhealthy, but some nutritionists believe that the type of saturated fat is also important. Stearic acid, a long-chain fatty acid consisting of 18 carbon atoms without double bonds is present in fairly constant proportions in beef, pork, lamb and veal (approxi-mately 9%–12% of total fatty acid content), with lower proportions found in poultry (approximately 6%–7% of total fatty acid content). It is biochemically classified as a saturated fatty acid for the purposes of food labeling and dietary recommendations[5].
The behavior of stearic acid is especially unique with respect to its effects on serum cholesterol levels. Studies in humans and experimental animals have suggested that ingestion of stearic acid has a neutral or cholesterol-lowering effect, which is in contrast to the effects of lauric, myristic and palmitic acids[6]. In addition, a beneficial effect of stearic acid on clotting factors can result in a less thrombogenic state. However, the molecular and cellular mechanisms by which it exerts these effects are still unclear[7].
Recently, it has been found that several fatty acids can function as natural ligands of peroxisome proliferator-activated receptors (PPARs) to regulate lipid homeostasis. PPAR are members of the nuclear hormone receptor family of ligand-dependent transcription factors. The PPAR subfamily comprises 3 isotypes: PPAR-α, PPAR-β, and PPAR-γ. Although a precise biological role for PPAR-β remains unclear, PPAR-α and PPAR-γ have modulatory roles in various cellular responses, including immune and inflammatory responses. Various fatty acids can regulate gene expression at micromolar concentrations through direct interactions with PPAR-α or PPAR-γ. Recently, it has been reported that ligands of PPAR-α or PPAR-γ can significantly protect the brain from ischemic or oxidative insult[8,9]. Stearic acid can function as one of these endogenous PPAR-α ligands[10]. Therefore, we hypothesize that stearic acid may function as a natural ligand of PPAR to protect neurons from ischemic insult.
Many studies have shown that ischemia-reperfusion injury (I/R injury) to the developing brain contributed to the neurodevelopmental handicaps suffered by infants who have experienced perinatal brain injury. Energy failure, excitatory amino acid release, and oxidative stress all contribute to injury in the cortex, striatum and hippocampus[11]. Therefore, on the basis of the most common causes of cerebral injury, we established 3 different injury models in vitro to screen a putatively neuroprotective fatty acid. In the present study, we evaluated the effects of stearic acid on brain slice injury induced by oxygen-glucose deprivation (OGD), glutamate or NaN3.
Materials and methods
Experimental animals Male Sprague-Dawley (SD) rats (50±5 g, grade II, certification N
Reagents and drugs Bisphenol A diglycidyl ether (BADGE), {3-[1(p-chlorobenzyl)-5-(isopropyl)-3-t-butyl-thiondol-2-yl]-2,2-dimethylpropanoic acid, Na} (MK886) and 2,3,5-triphenyltetrazolium chloride (TTC) were purchased from Sigma (St Louis, Missouri, USA). Stearic acid (Chemical Reagent Co Ltd, China) and all other reagents were of analytical grade. Stearic acid dissolved in ethanol was diluted in artificial cerebrospinal fluid (ASCF) before use. The final ethanol concentration in ASCF was less than 0.5%.
Brain slice preparation and drug exposure The slices were prepared as described by Gray et al[12] with several modifications. Whole brains were quickly removed and transferred to iced ACSF, which had the following composition (in mmol/L): NaCl 119, KCl 2.5, CaCl2 2, MgSO4 1, NaH2PO4 1.25, NaHCO3 26.2, glucose 10 (final pH 7.4). Brains were cut coronally into 400-μm thick sections with a vibrating tissue slicer (ZQP-86, Xiangshan, Zhejiang, China). Cortical and hippocampal slices were quickly isolated from the appropriate sections. Before being transferred to an experimental chamber, all slices were incubated in ACSF bubbled with 95% O2 and 5% CO2 at 32–34 °C for 90 min recovery. After full recovery, hippocampal slices were taken randomly into experimental chambers perfused with ACSF. The excitation electrode was planted in Shaffer collateral pathways of hippocampal slices under a microscope and a recording electrode was planted in the CA1 region. Population spikes (PS) were recorded to monitor tissue activity before the experi-ments. Slices were incubated with stearic acid, MK886 or BADGE in oxygenated ACSF for 3 h, and then the activities of the slices were evaluated by measuring the amplitude of the PS and using the TTC staining method.
OGD injury model Brain slices were transferred to experimental chambers and randomly assigned to 1 of 3 groups: (i) control group, in which slices were immersed in oxygenated ACSF at 34 °C; (ii) OGD group, in which slices were made anoxic by re-placing the ACSF with glucose-free ACSF equilibrated with 95% N2/5% CO2. After 10 min insult, the slices were re-oxygenated in ACSF for 2 h; (iii) OGD+stearic acid group, in which slices were incubated with different concentrations of stearic acid (3–30 μmol/L) 30 min prior to and during OGD insult.
Glutamate injury model Brain slices were transferred to experimental chambers and randomly assigned to 1 of 3 groups: (i) control group, in which slices were immersed in oxygenated ACSF at 34 °C; (ii) glutamate group, in which slices were subjected to 1 mmol/L glutamate with magnesium-free artificial cerebrospinal fluid for 30 min which had the following composition (in mmol/L): NaCl 143, KCl 5.4, CaCl2 1.8, NaH2PO4 1.0, N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES) 2.4, glucose 5.6, pH 7.4; (iii) glutamate+stearic acid group, in which slices were incubated with different concentrations of stearic acid (3–30 µmol/L) 30 min prior to and during glutamate application.
NaN3-induced injury model Brain slices were transferred to experimental chambers and randomly assigned to 1 of 3 groups: (i) control group, in which slices were immersed in oxygenated ACSF at 34 °C; (ii) NaN3 group, in which slices were subjected to 10 mmol/L NaN3 for 30 min; (iii) NaN3+stearic acid group, in which slices were incubated with different concentrations of stearic acid (3–30 µmol/L) 30 min prior to and during NaN3 application.
TTC staining After insult, slices were immersed in 2% TTC solution in a covered water bath shaker at 37 °C for 1 h and wet weight was measured after the slices were rinsed twice with saline. An extracting solution (50:50 mixture of ethanol/dimethylsulfoxide) was added to the vials of slices at a rate of 20 mL per 1 g of slice. The extracted liquid was added to 96-well plates (200 µL per well) for 24 h and the optical density of each well was measured at 490 nm (OD490) by using an enzyme-linked immunosorbent assay (ELISA) reader (Bio-TEK Elx800uv)[13].
Blockade of the effect of stearic acid by PPAR inhibitors MK886 is an indole compound that was originally identified as a potent inhibitor of 5-lipoxygenase (5-LOX)-activating protein (FLAP). Recently, it has been found to inhibit PPAR-α via a non-competitive mechanism as shown by its effects on the binding of arachidonic acid to PPAR-α protein. Brain slices were treated with 5 µmol/L MK886 or 100 µmol/L BADGE (PPAR-γ antagonist) in the presence or absence of stearic acid to determine whether antagonists of PPAR could block the neuroprotection provided by 30 µmol/L stearic acid. The tissue activities were measured by using the TTC method.
Statistical analysis Four to six brain slices from 3 rats were used for each experimental group, and data collected from 6 independent experiments were used to calculate means, which are expressed as mean±SD. SPSS statistical software 10.0 for Windows was used and statistical significance was evaluated by using one-way ANOVA and the SNK test. Statistical significance was assumed if P<0.05.
Results
Brain slice preparation and drug exposure The hippocampus is very sensitive to hypoxic and chemical hypoxia. Hypoxia and tissue injury are both associated with a decreased amplitude of or disappearance of population spikes (Figure 1).
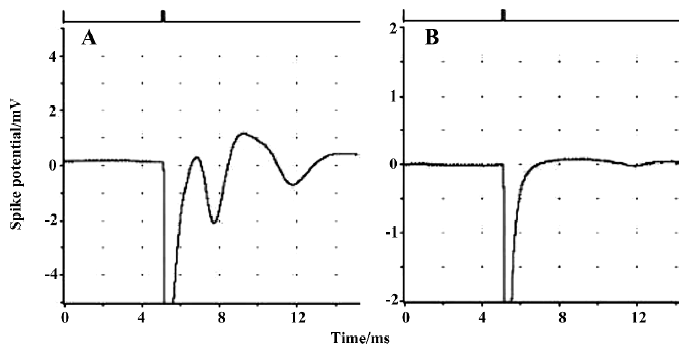
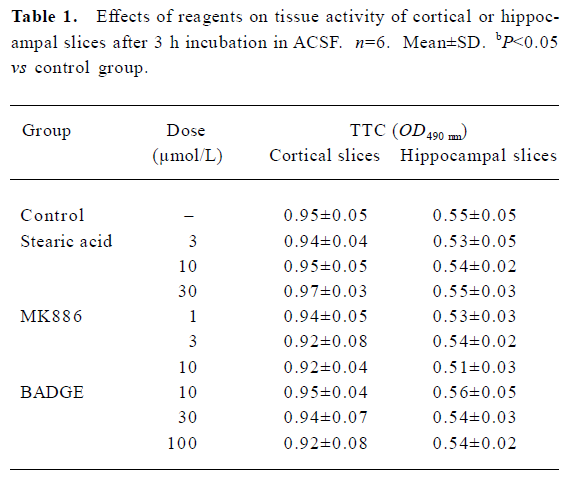
Population spikes (PS) in CA1 regions of randomly selected hippocampal slices indicated the recovery of brain slices after incubation in ACSF for 90 min. These recovered slices were used for the subsequent experiments. None of the brain slices exhibited significant tissue damage (P>0.05) after being incubated with different concentrations of stearic acid (3–30 µmol/L), MK886 (1–10 µmol/L) or BADGE (10–100 µmol/L) in ACSF for 3 h (as shown in Table 1). Based on their IC50 values and our screening results, we chose concentrations of reagents that did not produce significant effects on tissue activity for the experiments described below.
Full table
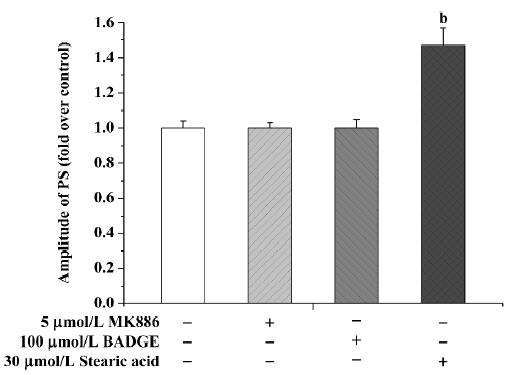
Stearic acid at a concentration of 30 µmol/L induced a significant increase in PS amplitude, reaching 145%±10% (P<0.05) of basal level after 3 h treatment. Because stearic acid may have interfered with amplitude of PS, we only used the TTC staining method to assess tissue activity. Neither MK886 nor BADGE affected the PS amplitude after 3 h incubation (P>0.05; Figure 2).
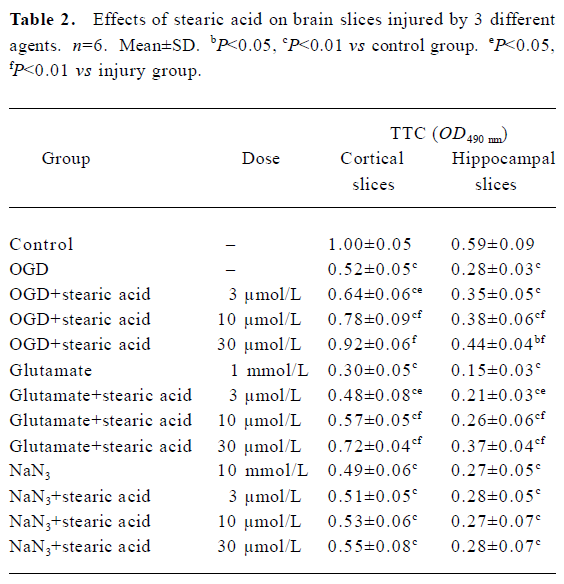
Brain slice insult with OGD After they were subjected to 10 min of OGD insult and 2 h of post-incubation, the activities of the brain slices were reduced by approximately 50%. There was a statistically significant difference between the OGD and control groups (P<0.01). Stearic acid can dose-dependently protect against the decreases in tissue activity induced by OGD insult (Table 2).
Full table
Brain slice insult with glutamate Only approximately 30% of tissue activity remained after brain slices were incubated with 1 mmol/L glutamate for 30 min. There was a statistically significant difference between the injury and control groups (P<0.01). Stearic acid can dose-dependently protect against the decreases in tissue activity induced by glutamate insult (Table 2).
Brain slice insult with NaN3 After they were subjected to 30 min exposure to NaN3, the activities of the brain slices were reduced by approximately 50%. There was a statistically significant difference between the NaN3 and control groups (P<0.01). Stearic acid did not protect brain slices from NaN3 insult (P>0.05; Table 2).
Blockade of the effects of stearic acid by PPAR inhibitors It is known that 5-LOX inhibitor can protect against ischemic-like injury in PC12 cells in vitro by modulating p38 MAP kinase activity[14]. Therefore, we tested whether MK886 or BADGE could directly protect brain slices against OGD or glutamate insult. MK886 at a concentration of 5 µmol/L significantly improved the activity in tissue insulted by OGD or glutamate (P<0.05), whereas BADGE at a concentration of 100 µmol/L produced no significant effect on the activity of tissue insulted by OGD or glutamate (P>0.05). When slices were treated with 5 µmol/L MK886 during pre-incubation, the protection against OGD or glutamate insult afforded by stearic acid was not abolished. In contrast, 100 µmol/L BADGE blocked its protective effect completely (Table 3).
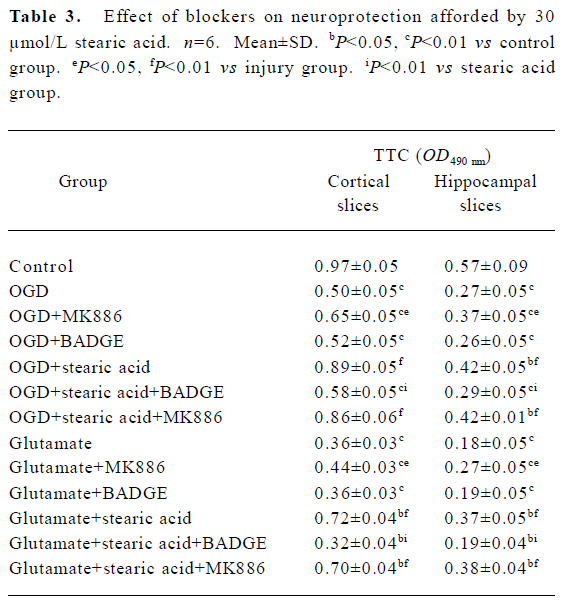
Full table
Discussion
Experiments using brain slices have the advantages of in vivo and in vitro studies: they not only maintain anatomic relations and natural synaptic connectivity in vitro, but also eliminate such in vivo variables as blood flow, temperature and ionic environment, and closely match in vivo conditions. Therefore, increasing numbers of brain slice models have been used to study brain function and brain protection. In the present study, we used 3 different damage models to reflect the pathological characteristics of different phases of I/R injury (metabolism disorder, toxic amino acid and oxidative stress)[11]. The present study is the first to demonstrate that stearic acid can dose-dependently protect rat brain slices against OGD and glutamate toxicity, but not against NaN3 toxicity.
As an early consequence of OGD associated with brain ischemia, neuronal aerobic metabolism and ATP production are severely influenced. The decrease in energy production and malfunction of Na+/K+-ATPase leads to loss of active ion transport, destruction of transmembrane electrochemical ionic gradients and membrane depolarization, which promotes presynaptic glutamate release and impaired uptake [11]. Because brain does not rely on stearic acid oxidative metabolism for the production of ATP, the neuroprotective effects provided by stearic acid against OGD insult may result from the blockage of excitatory amino acid (EAA) receptors, defense against oxidative stress, presynaptic depression of glutamate release or promotion of glutamate uptake[11]. It is known that the presence of 0.5 mmol/L glutamate in the extracellular space can reverse glutamate uptake and decrease the synthesis of glutathione in neurons[15]. In the present study, 1 mmol/L glutamate was applied to induce brain injury, and stearic acid was found to dose-dependently protect brain slices from glutamate insult. Our results suggest that the neuroprotective effects of stearic acid are related to the blockage of EAA receptors and/or defense against oxidative stress.
Oxidative stress is a particularly important factor in mitochondrial dysfunction because the respiratory chain continually leaks superoxide free radicals. These reactive oxygen species lead to generalized oxidative damage to all mitochondrial components. Mitochondria are the powerhouse of the cell: their primary physiological function is to generate ATP through oxidative phosphorylation via the electron transport chain. NaN3, an inhibitor of cytochrome c oxidase (complex IV of the mitochondrial electron transfer chain), significantly decreased the tissue activity of brain slices in the present study. In our experiments, stearic acid did not protect brain slices against NaN3 insult, which suggests that it acts after the malfunction of the mitochondrial electron transfer chain during ischemia.
In some experiments, different ligands have been used according to the different isotypes of PPAR[16–18], and these agonists all play a role in neuroprotection to different extents. Several fatty acids bind to all 3 PPAR isoforms, although there is a preference of PPAR-α for polyunsaturated fatty acids. MK886 and BADGE are 2 important antagonists of PPAR. We found that BADGE completely blocked the neuroprotective effect provided by stearic acid, whereas MK886 did not abolish its protective effect. MK886 can directly protect brain slices against OGD or glutamate insults. Although MK886 did not abolish the effects of stearic acid, we cannot conclude that PPAR-α is not involved in the process of neuroprotection. However, BADGE could completely block the neuroprotective effect of stearic acid, which suggests that PPAR-γ may play a more important role in the protective effect, because BADGE has no significant effect on injured brain slices by itself. It has been reported that stearic acid 200 µmol/L could only enhance reporter gene expression under the control of ideal PPAR responsive element (PPRE
The findings from the present study suggest that stearic acid can effectively protect brain during the early stages of I/R in immature rats. Newborn animals are more tolerant than adults of cerebral hypoxia/ischemia insult, and their blood-brain barriers are easier to pass. Stearic acid may contribute to protection against hypoxic-ischemic brain damage in the immature brain.
In conclusion, stearic acid can protect brain slices (cortical and hippocampal) against injury induced by OGD or glutamate. Its neuroprotective effect is possibly mainly mediated by the activation of PPAR-γ in brain tissue.
References
- McArthur MJ, Atshaves BP, Frolov A, Foxworth WD, Kier AB, Schroeder F. Cellular uptake and intracellular trafficking of long chain fatty acids. J Lipid Res 1999;40:1371-83.
- Needleman P, Raz A, Minkes MS, Ferrendelli JA, Sprecher H. Triene prostaglandins: prostacyclin and thromboxane biosynthesis and unique biological properties. Proc Natl Acad Sci USA 1979;76:944-8.
- Bazan NG. Lipid signaling in neural plasticity, brain repair, and neuroprotection. Mol Neurobiol 2005;32:89-103.
- Hwang DH. Essential fatty acid and immune response. FASEB J 1989;3:2052-60.
- Baer DJ, Judd JT, Kris-Etherton PM, Zhao G, Emken EA. Stearic acid absorption and its metabolizable energy value are minimally lower than those of other fatty acids in healthy men fed mixed diets. J Nutr 2003;133:4129-34.
- Monsma CC, Ney DM. Interrelationship of stearic acid content and triacylglycerol composition of lard, beef tallow and cocoa butter in rats. Lipids 1993;28:28539-47.
- Tholstrup T, Marckmann P, Jespersen J, Sandstrom B. Fat high in stearic acid favorably affects blood lipids and factor VII coagulant activity in comparison with fats high in palmitic acid or high in myristic and lauric acids. Am J Clin Nutr 1994;59:371-7.
- Brune S, Kolsch H, Ptok U, Majores M, Schulz A, Schlosser R, et al. Polymorphism in the peroxisome proliferators activated receptor alpha gene influences the risk for Alzheimer’s disease. J Neural Transm 2003;110:1041-50.
- Colville-Nash PR, Qureshi SS, Willis D, Willoughby DA. Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: correlation with induction of heme oxygenase 1. J Immunol 1998;161:978-84.
- Ehrmann J Jr, Vavrusova N, Collan Y, Kolar Z. Peroxisome proliferator-activated receptors (PPARs) in health and disease. Biomed Papers 2002;146:11-4.
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev 1999;79:1431-68.
- Gray JJ, Bickler PE, Fahlman CS, Zhan X, Schuyler JA. Isoflurane neuroprotection in hypoxic hippocampal slice cultures involves increases in intracellular Ca2+ and mitogen-activated protein kinases. Anesthesiology 2005;102:606-15.
- Preston E, Webster J. Spectrophotometric measurement of experimental brain injury. J Neurosci Methods 2000;94:187-92.
- Song Y, Wei EQ, Zhang WP, Zhang L, Liu JR, Chen Z. Minocycline protects PC12 cells from ischemic-like injury and inhibits 5-lipoxygenase activation. Neuroreport 2004;15:2181-4.
- Szatkowski M, Attwell D. Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci 1994;17:359-65.
- Simonin MA, Bordji K, Boyault S, Bianchi A, Gouze E, Bécuwe P, et al. PPAR-γ ligands modulate effects of LPS in stimulated rat synovial fibroblasts. Am J Physiol Cell Physiol 2002;282:125-33.
- Kwak BR, Myit S, Mulhaupt F, Veillard N, Rufer N, Roosnek E, et al. PPAR-γ but not PPAR-α ligands are potent repressors of major histocompatibility complex class II induction in atheroma-associated cells. Circ Res 2002;90:356-62.
- Wayman NS, Hattori Y, Mcdonald MC, Filipe HM, Cuzzocreas S, Pisano B, et al. Ligands of the peroxisome proliferator-activated receptors (PPAR-γ and PPAR-α) reduce myocardial infarct size. FASEB J 2002;16:1027-40.
- Wolfrum C, Borrmann CM, Börchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors α- and γ-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci USA 2001;98:2323-8.