
Establishment of multiplexed, microsphere-based flow cytometric assay for multiple human tumor markers1
Introduction
Tumors, especially the malignant tumor, are still one of the leading causes of death in the world. Early screening and detection of suspected cancer patients is the key to improving the therapeutic effect. However, early diagnosis is difficult because clinical presentations are highly variable and signs are often subtle and common to a variety of conditions. A tumor marker is a substance sometimes found in an increased amount in the blood, other body fluids, or tissues and may mean that a certain type of cancer is in the body. The screening test by tumor marker has been proven to be an effective method for detecting cancer in asymptomatic individuals[1,2]. An ideal tumor marker would be of 100% sensitivity and specificity, reflect the change in tumor burden, allow for therapeutic intervention, and predict prognosis and recurrence. Unfortunately, the tumor markers available nowadays can not meet all these characteristics. For example, only 50%–70% of hepatocellular carcinoma patients showed a significant enhancement in the amount of alpha-fetoprotein (AFP) in sera. However, the use of panels with 2 or more approved markers can contribute to the improved sensitivity and diagnostic efficiency. Therefore, an analysis of the complete set of tumor markers is often of more value than an analysis of a single isolated marker.
Recent advances concerning the applications for the simultaneous detection of various molecules have resulted in different microsphere-based flow cytometric assays (MFCA). The MFCA developed by Luminex (Austin, TX, USA) involves the covalent coupling of a capture antibody or target molecule on polystyrene microspheres (5.6 µm). The microspheres are internally dyed with red and orange fluoro-phores. By varying the ratio of the 2 fluorophores, up to 100 different bead sets can be distinguished and each bead set can be coupled to a different biological probe. A reporter molecule labeled with a fluorescent marker (such as phycoerythrin [PE] or Alexa-532) binds to the analyte captured on the beads. The bound microspheres are passed though a flow cytometer specially designed to identify the different microsphere sets. Quantification is based on the intensity of the fluorescent reporter signal. Previous studies have used this type of technology for multiplexed assays of cytokines, antibodies, hormones, nucleic acids, viruses, and other biomolecules[3,4]. However, at present, either the specific antibody pairs or the availability of predefined kits limits these assays. In addition, earlier investigations did not report the reproducibility of the protein-microsphere coupling procedure, which is crucial for long-term, particle-based flow cytometric assays.
To overcome these limitations, in the present study we chose to develop and validate our own MFCA to quantify tumor markers as a means to simultaneously measure the levels of human AFP, carcinoembryonic antigen (CEA), cancer antigen (CA)19-9, CA24-2, and CA72-4. We chose these 5 common abdominal tumor markers in order to screen and detect suspected cancer patients. We compared the performance of MFCA with that of a conventional ELISA system. Specifically, we determined whether the imprecision in reproducing the coupling of capture antibodies to beads at different times affected the results obtained in assays of human tumor markers. We also compared the upper and lower limits for the detection of human tumor markers using MFCA and ELISA. Finally, we compared the reproducibility of MFCA for measuring tumor markers in human serum samples. We showed that MFCA is a reliable, fast, and reproducible technique with a sensitivity that is comparable to that of conventional ELISA.
Materials and methods
Serum sample preparation Blood samples were collected from 10 patients with abdominal tumors, including 4 patients with hepatocellular carcinoma, 4 patients with colon cancer, 1 patient with pancreatic carcinoma, and 1 patient with cholangiocarcinoma at the initial diagnosis of the diseases, as well as 4 healthy, adult control donors. All participants of this study signed an informed consent approved by the Institutional Review Board. Blood samples (5 mL) were collected in glass tubes without additives and allowed to clot at room temperature for 60 min. The serum was separated by centrifugation at 3000×g for 30 min. Aliquots of serum (150 µL) were taken and stored at -80 °C until ready for use. The time from collection to frozen storage was no more than 3 h. The samples were collected and contained no identifying features that would make it possible for the investigators participating in the study to identify the patients.
Reagents Carboxylated polystyrene microspheres numbers 101, 103, 105, 107, and 109, each with a distinct emitting fluorescence pattern, were purchased from Luminex (USA). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (sulfo-NHS) were provided by Pierce (Rockford, IL, USA). 2-(N-Morpholino))ethanesulfonic acid (MES), streptavidin-R-PE (SA-PE), streptavidin-Alexa-532, and streptavidin-horseradish peroxidase were from Sigma (St Louis, MO, USA). PE-conjugated goat antimouse immunoglobulin G (IgG) was supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). The DSB-X biotin protein labeling kit was provided by Molecular Probes (Eugene, OR, USA).
Human AFP, CEA, CA19-9, CA24-2, and CA72-4 mouse monoclonal antibody pairs, directed against different non-competing epitopes of their respective targets, and standard recombinant proteins were purchased from Biodesign (Saco, ME,USA). First, azide, tris, glycine, or other nitrogen-containing compounds were removed by dialysis overnight in phosphate-buffered saline (PBS) using Slide-A-Lyzer cassettes (Pierce, USA) since these substances can interfere with the coupling reaction. All recombinant proteins were reconstituted in PBS (pH 7.4) containing 1% bovine serum albumin (BSA) to a concentration of 10 μg/mL. All proteins were aliquoted and stored at -20 °C.
Covalent coupling of capture antibodies to fluorescent microspheres Covalent coupling was performed by following the protocols recommended by Luminex with some modifications[5,6]. In short, after being washed twice with 200 µL activation buffer (0.1 mol/L NaH2PO4 [pH 6.2]), spectrally differentiable carboxylated microspheres (2.5×106) were pelleted (10 000×g for 5 min) in 1.5 mL Eppendorf tubes in a microcen-trifuge (Eppendorf, Hamburg, Germany). The microspheres were resuspended by sonication (30 s at 1120 W, 50–60 Hz) and gentle vortexing (VWR International, West Chester, PA, USA) in 80 µL activation buffer to which 10 µL of each solution of sulfo-NHS and EDC (both at 50 mg/mL), prepared in ddH2O immediately before use, was sequentially added to stabilize the reaction and activate the microspheres. The suspension was allowed to incubate for 20 min at room temperature and then resuspended in 500 µL coupling buffer [0.05 mol/L MES (pH 5.0)] containing 125 µg capture antibody. The mixture was incubated for 2 h in the dark with continuous shaking. The 250 µg/mL concentration of the antibody was used for coupling because our preliminary experiments indicated that this coupling concentration yielded high median fluorescent intensities (MFI) and good inhibition (data not shown). The coupled microspheres were then incubated in 500 µL blocking buffer [PBS-TBN: PBS containing 0.1% BSA, 0.02% Tween-20, and 0.05% NaN3 [pH 7.4)] for 30 min to block the uncoupled sites. An aliquot of the microspheres was diluted at 1:5 in PBS-TBN to count 2 of the 9 large squares on the grid on the hemacytometer (Bright Line, VWR International, West Chester, PA, USA). The concentration was calculated as follow: Bead count/2 (N
Testing the efficiency and density of antibody coupling to the microspheres After prewetting the 1.5 mL Eppendorf tube with assay buffer (PBS containing 1% BSA [pH 7.4]), 50 µL of the antibody-coupled microsphere sets (at a final concentration of 100 microspheres of each set/µL) and 50 µL PE-conjugated goat antimouse IgG detection antibody (2-fold serial dilution from 4 to 0.0625 µg/mL) were added into the tube. After incubation for 30 min and washing twice with PBS-1% BSA, the microspheres were measured and analyzed with the Luminex 100 system. The capture antibody density was measured by computing the ratio of the MFI of the coupled and uncoupled beads that reacted with the PE-conjugated goat antimouse IgG secondary antibody.
Detection of non-specific cross-reactivity in multiplexed MFCA The biotin labeling of the detection antibody was performed by following the commercial kit specifications. To detect whether non-specific cross-reactivity between components during the assay occurred, and consequently, to monitor false positive findings, a full microsphere mixture with the biotinylated detection antibodies for each microsphere were incubated in the presence of a single tumor marker standard at 64 µg/mL (AFP and CEA) or 64 U/mL (CA19-9, CA24-2, and CA72-4). After incubation for 30 min and washing twice with PBS-1% BSA, the microspheres were measured and analyzed with the Luminex 100 system.
Multiplex tumor marker assays The establishment of the standard curve of MFCA was prepared with 4-fold serial dilution steps (from 64 to 0.001 µg/mL for AFP and CEA or from 64 to 0.001 U/mL for CA19-9, CA24-2, and CA72-4) of recombinant protein standards of tumor markers. Each standard was tested in a single bead assay to determine the optimal concentration of the detection antibody. Next, the microspheres were multiplexed and optimized for the incubation times and reporter signal. As a reporter signal, both streptavidin-Alexa-532 and SA-PE were tested in different concentrations. The samples were measured twice and blank values were subtracted from all readings. All assays were carried out directly in a 1.5 mL Eppendorf tube at room temperature and protected from light. A mixture containing 5000 microspheres/set was incubated together with a cocktail of biotinylated antibodies and a standard, sample, or blank of the indicated concentration in a final volume of 100 µL for 30 min under continuous shaking. The microspheres were then washed twice with PBS-1% BSA in order to remove any unbound antibodies. After 30 min of incubation with SA-PE and washing twice with PBS-1% BSA again, the microspheres were measured in a final volume of 100 µL and counted 50 µL and total 50 of each set of microspheres to obtain the MFI. The analyzer contains 2 solid state lasers: a classification laser (635 nm, 10 mW maximum) excites the fluorochromes impregnated within the microsphere, and a reporter laser (532 nm, 100 mW maximum) excites fluorescent molecules bound to the microsphere surface. The classification emission spectrum does not overlap with the reporter emission signal. Therefore, compensation is not necessary as with a conventional flow cytometer.
Tumor markers ELISA ELISA was performed by coating 96-well polystyrene plates (Corning, Corning, NY, USA) with a specific monoclonal antibody. Standards and samples were added to the appropriate wells after the plates were blocked with non-fat, dry milk, and the plates were incubated overnight at 4 °C. A specific biotinylated antibody was then added to all the wells after they were washed with PBS, and they were incubated for 1 h at room temperature. The plates were washed with PBS again and incubated for another 30 min with horseradish peroxidase-conjugated streptavidin. After the removal of the non-bound horseradish peroxidase conjugate by washing, tetramethylbenzidine dihydrochloride substrate reagent solution (ICN Biomedicals, Aurora, OH, USA) was added to the wells. The reaction was stopped with 25 µL/well of 2 mol/L H2SO4. The plates were read at 450 nm with a microplate spectrophotometer (Molecular Devices, Menlo Park, CA, USA) and the results were analyzed with associated software.
Results
Optimization of reagents and procedures Specific antibody pairs were used to develop and validate this multiple human tumor marker assay. The critical parameters of this assay are the choice of primary antibodies, the concentration of the biotinylated detection antibodies, the amount and choice of conjugated fluorochromes, and the incubation times of the samples and the conjugated fluorochromes. Several commercial antibody pairs suitable for use in ELISA might not perform well in our multiplex system; either the coupling to the microspheres would be ineffective or the beads would fail to report a signal from the recombinant standards. The test for the coupling efficiency (such as AFP) showed that the highest coupling readings were approximately 15 000 MFI, while backgrounds were lower than 100 MFI, which proved that antibody coupling to the microspheres was successful (Figure 1).
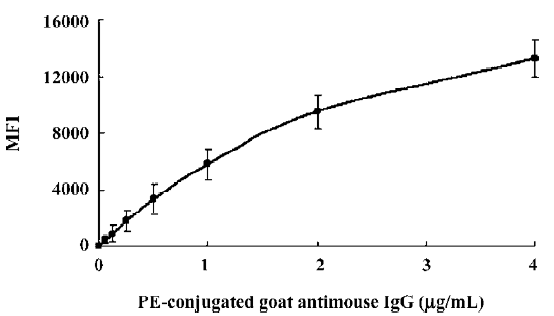
The biotin-conjugated detection antibodies were optimized in a single bead assay before they were added to the multiplex profile. The amount of the conjugated antibody used varied per tumor marker and the optimal concentrations of detection antibody for this assay ranged between 1.0 and 5.0 µg/mL. As a reporter signal, both streptavidin-Alexa-532 and SA-PE were tested in different concentrations ranging from 0.1 to 1.0 µg/mL for Alexa-532 and 0.1 to 5.0 µg/mL for SA-PE in the present study. Alexa-532 turned out to be less suitable for several of our antibodies because we were unable to pick up a signal at a different concentration of fluorochromes, whereas SA-PE resulted in a signal. Therefore, we used SA-PE for further experiments and the optimal concentration was 4 µg/mL. Prolonging the incubation times for either the sample or SA-PE did not improve our ability to detect any tumor markers. Overall MFI signals increased with longer incubation times, but sensitivity was lost due to a proportionally higher increase in the background signal.
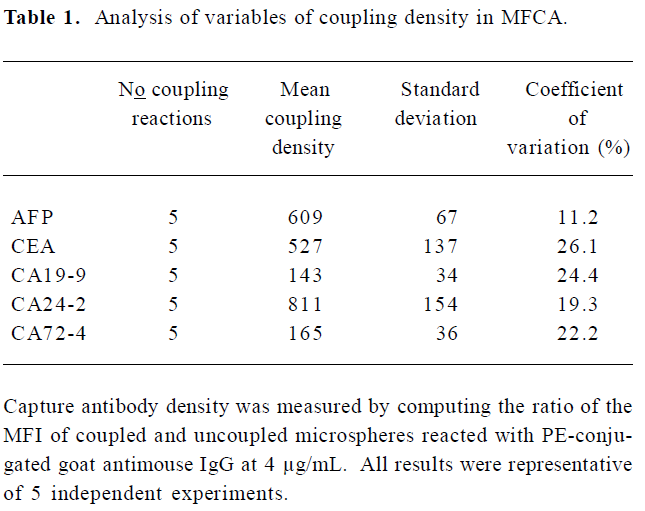
Variability of antibody-bead coupling and its effect on assays For long-term studies, it is crucial that the process of coupling the capture antibody to beads can be reproduced so that bead sets coupled independently will give consistent assay results. To address this concern, we tested how the density of capture antibodies on the beads varied during repeated coupling procedures. We observed that minor variations in the coupling procedures could markedly affect the density of coupled capture antibodies. Table 1 indicates the variation of the coupled antibody density of the beads after 5 independent coupling reactions. The coefficient of variation for the density of the independently coupled bead sets ranged from 11.2% (AFP) to 26.1% (CEA). In subsequent studies, we found that the optimal concentrations of the capture antibodies for coupling were generally those that resulted in the greatest density of antibodies coupled to beads. Beads with high coupling densities tended to give more sensitive assays than beads with low coupling densities (Figure 2).
Full table
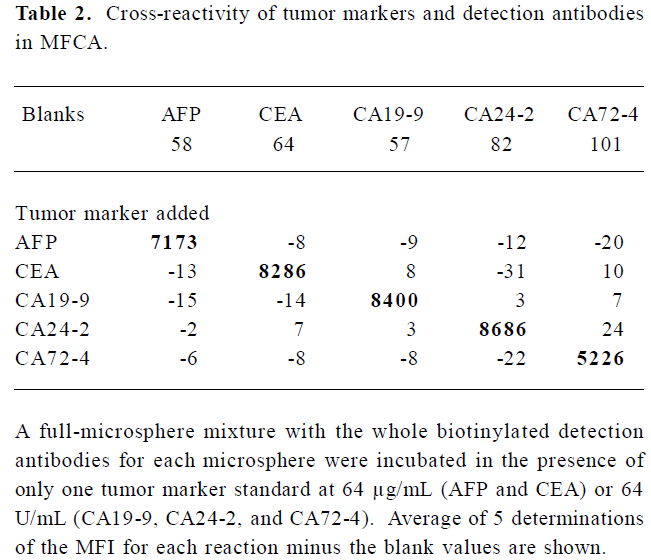
Identification of cross-reactivity As shown in Table 2, although the background varied with each microsphere, none of the specific antibody sets gave readings above background with non-relevant tumor marker standards, indicating that there was no detectable cross-reactivity. Positive readings were found for the microspheres labeled with the specific capture antibody.
Full table
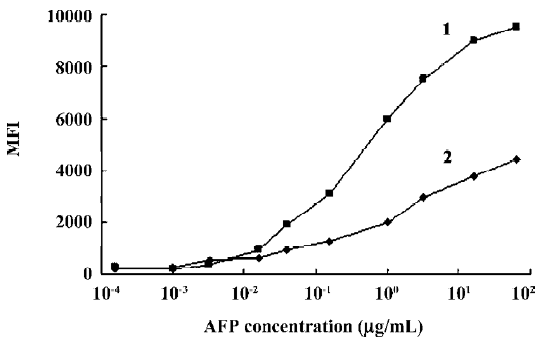
Range of detection for standard curves The standard curves for all 5 tumor markers are represented in Figure 3. We compared the lower, upper, and total working detection ranges for ELISA and MFCA single and multiplexed assays of these 5 tumor marker standards. The approximate upper and lower limits of detection were conservatively estimated by comparing the largely linear regions of the standard curves in which fluorescence and absorbance were plotted linearly and concentration logarithmically. We excluded the uppermost region of the multiplexed MFCA where the slopes differed markedly from those of the individual assays. As shown in Table 3, ELISA had lower limits of detection compared to MFCA. For each assay, ELISA was more sensitive by a factor of 4–16. Conversely, MFCA had a higher detection limit by a factor of 4–64. The total dynamic ranges were greater for MFCA than ELISA. In addition, we did not find that multiplexed MFCA were less sensitive than the individual assays.
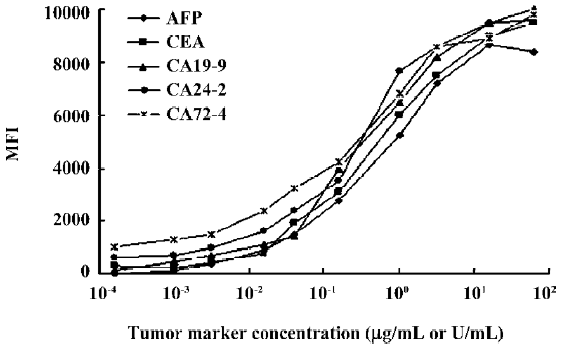
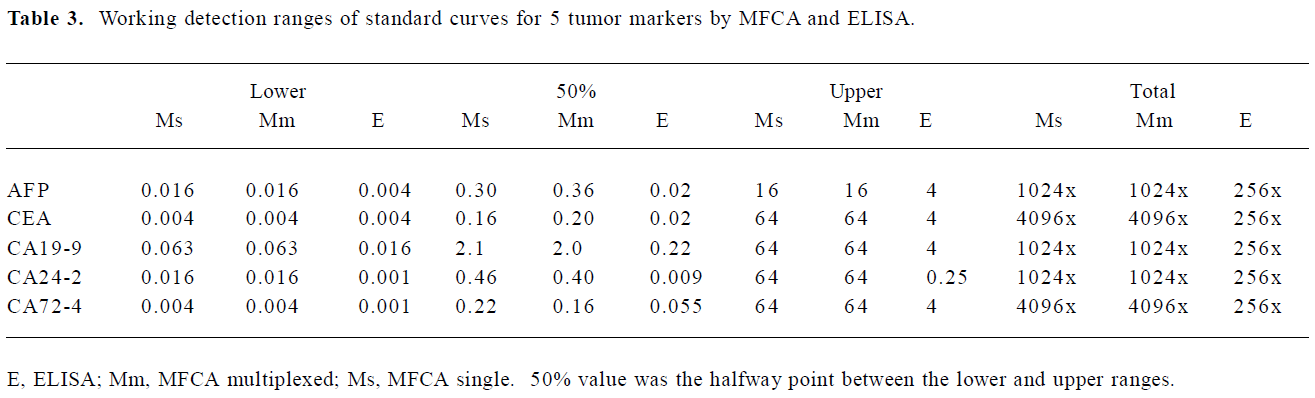
Full table
Reproducibility of MFCA in serum samples The results of MFCA were compared with those of ELISA for serum samples from healthy individuals and tumor patients with an unknown concentration of each tumor marker, which would be widely disparate at the linear part of the standard curves of the multiplex assay. Correlations between both techniques were determined by measuring the levels of all tumor markers with the MFCA system and with conventional ELISA. All the samples measured with the MFCA system were assayed twice and at different time points so that the intra-assay variance could be determined.
The concentrations of the samples measured with ELISA concurred with the concentrations that were determined with the MFCA system (for example, AFP in Figure 4). High correlation coefficients (r2), ranging from 0.927 to 0.985, were found for all the tumor markers. Slopes varied between 0.86 and 1.03, with a mean slope of 0.95. Intra-assay variability, expressed as a coefficient of variation, was calculated based on the average for 14 serum samples and measured twice in the multiplex assay repeated at 2 different time points. The intra-assay variability within the replicates of the samples was comparable to that of ELISA (<10%) with an average coefficient of variation of 8.92%. Inter-assay variability was evaluated by testing quadruplicates of 1 standard (1 µg/mL for AFP and CEA or 1 U/mL for CA19-9, CA24-2, and CA72-4) that was positioned at the upper linear part of the standard curve of all the tumor markers in a multiplex assay at 4 different time points. The variabilities of these samples were between 10.4% and 18.6%, with an average of 15.06% (Table 4).
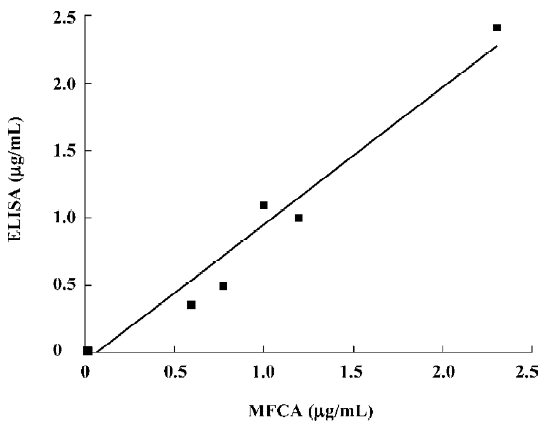
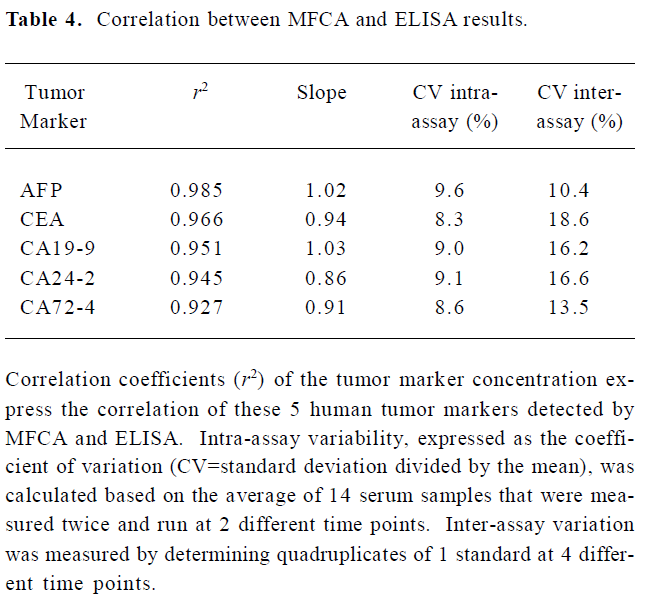
Full table
Discussion
Tumor markers are soluble proteins that are secreted by tumor cells or other normal cells stimulated by the tumor microenvironment and can reflect the existence of a type of tumor in the body before the presentations of obvious symptoms. It is at this early stage that most cancer patients will have the greatest chance of cure. Immunoassays, such as ELISA, can provide relatively sensitive, specific, and precise quantitation of tumor markers, but ELISA can detect only one analyte in a single test, thus, when applied to the expanding number of tumor markers that are involved in defining a particular system, they become expensive and time consuming to perform. In addition, ELISA has more limitations, like the need for a large sample volume, the narrow dynamic range, and complicated dilution procedures[7,8]. A protein microarray can analyze thousands of proteins simultaneously, but it can not quantify accurately[9]. As we know, a test of tumor markers is not simply positive or negative and it is of great clinical significance when exceeding a given threshold. Continued technological advances are expected to provide not only new analyte markers, but also new technologies to detect them. Here we reported the establishment of the MFCA system for the detection of multiple human tumor markers and the results showed that our multiplexed assay was comparable in sensitivity, accuracy, and reproducibility to the “gold standard” which was the ELISA.
The basic principle behind this technology involves microspheres being labeled with a distinguishable fluoro-phore that allows it to be assigned or gated to a particular region by the scanner. Capturing antibodies specific for the protein of interest is covalently linked to beads of a unique fluorescent region. The combination of different beads allows the user to simultaneously measure various proteins in the flow cytometer. From the immunological point of view, the limitation of this technique is the availability of matched antibody pairs that do not cross-react with other reagents. Numerous matched pairs of high-affinity and high-specificity antibodies have since been developed and commercialized, which encourage us to evaluate a multiplexed format of tumor markers. We have observed that minor alterations of the coupling process may significantly affect the density of coupled antibodies. Subsequent assays using bead sets with a low density of coupled antibodies tended to be less sensitive and reliable than that with relatively high density. Thus, it is important to check whether coupled microspheres have similar densities of capture reagents if repeating the coupling procedures and to optimize the concentration of antibodies coupled to beads.
In the present study, we have demonstrated that MFCA has a clear advantage over conventional ELISA, including the ability to detect large numbers of analytes simultaneously, therefore providing a powerful tool for profiling multiple tumor markers. Theoretically, this assay can detect up to 100 analytes in a single sample and thus more novel markers are readily welcome to be added in this open system. Consequently, the sample volume required to test a single marker by ELISA would be sufficient to detect all analytes by MFCA[10,11]. Obviously, this is a major advantage when working with a small sample. Furthermore, the MFCA took 2.5 h not including coupling process nor flow cytometric analysis. In contrast, ELISA took 5 h nor including overnight capture antibody incubation and the spectrophotometric analysis. Alternatively, some commercial kits containing bead sets already coupled to capture antibodies are available, and the variety of available kits continues to expand as this new technology grows. Such kits will eliminate the time-consuming coupling process and simplify the detection steps by using automated XY platform (XYP) plate handler.
In addition, MFCA is sensitive and accurate since this multiplex assay has been set up as a sandwich immunoassay, which will enhance specificity and reduce the risk of cross-recognition with other proteins. Moreover, each fluorescence signal is the mean of 50 measurements of a single microsphere, and each microsphere represents an assay by itself, whereas with ELISA, the samples are usually tested merely in duplicate or triplicate. Although higher numbers of events are commonly used in MFCA, using as few as 50 events will not affect the precision of the assay. Within a liquid phase, the chance of the interaction of proteins with their complementary antibodies will increase, which will lead to less non-specific binding and thus to lower background signals[12–14]. Unlike Carson et al[15], we did not find that multiplexed MFCA were less sensitive than the individual assays as evidenced by Table 3. However, we tested only 5 not 15 analytes simultaneously in this study, thus, our multiplexed assays were less likely to be adversely affected by antibody cross-reactivity and interference.
Moreover, each standard curve can be adjusted to a biological range in which a sample can be expected depending on the origin of the specimen. With this wide dynamic range of standard curves, samples do not have to be diluted or concentrated[16,17], but it is crucial to confirm that these parameters are achieved with high-affinity antibodies that define the lower end of sensitivity of the assay and the rapid reaction kinetics. Multiplexed assay performance is mainly dependent on the quality of the antibodies. Therefore, potential antibodies should be screened initially by comparing the slopes of single and multiplexed standard curves to detect possible cross-reactivity problems with the multiplexed reagents.
We have shown that MFCA can be used to generate quantitative biomarker data for serum samples, which appear to be more reproducible than ELISA. MFCA is less error prone because of its more direct fluorescence detection system as opposed to the more indirect detection method of ELISA, which is based on enzyme amplification. However, this multiplex assay involves mixing a large number of different antibodies in a single reaction. These antibodies could bind non-specifically to any other component within the assay and serve as antigens for other immunoglobulins, causing false positive values or blocking the readout. Thus, when sera are used as the matrix, the matrix has to be carefully monitored for blocking substances like heterophilic antibodies. In conclusion, MFCA provides a more rapid and less expensive method that has a 3–4 logarithmic range of sensitivity compared with 1–2 logs for ELISA, as well as the specificity and reproducibility expected from the latter[18]. Therefore, the successful establishment of the MFCA system for multiple human tumor markers provides the foundation for the further study of clinical applications.
References
- Sturgeon C. Practice guidelines for tumor marker use in the clinic. Clin Chem 2002;48:1151-9.
- Cillo U, Navaglia F, Vitale A, Molari A, Basso D, Bassanello M, et al. Clinical significance of alpha-fetoprotein mRNA in blood of patients with hepatocellular carcinoma. Clin Chim Acta 2004;374:129-38.
- Hulse RE, Kunkler PE, Fedynyshyn JP, Kraig RP. Optimization of multiplexed bead-based cytokine immunoassays for rat serum and brain tissue. J Neurosci Method 2004;136:87-98.
- Martins TB, Pasi BM, Litwin CM, Hill HR. Heterophile antibody interference in a multiplexed fluorescent microsphere immunoassay for quantitation of cytokines in human serum. Clin Diagn Lab Immunol 2004;11:325-9.
- Jager W, Velthuis H, Prakken BJ, Kuis W, Rijkers GT. Simultaneous detection of 15 human cytokines in a single sample of stimulated peripheral blood mononuclear cells. Clin Diagn Lab Immunol 2003;10:133-9.
- Tarnok A, Hambsch J, Chen R, Varro R. Cytometric bead array to measure six cytokines in twenty-five microliters of serum. Clin Chem 2003;49:1000-2.
- Jia XC, Raya R, Zhang L, Foord O, Walker WL, Gallo ML, et al. A novel method of multiplexed competitive antibody binning for the characterization of monoclonal antibodies. J Immunol Methods 2004;288:91-8.
- Biagini RE, Sammons DL, Smith JP, MacKenzie BA, Striley CA, Semenova V, et al. Comparison of a multiplexed fluorescent covalent microsphere immunoassay and an enzyme-linked immunosorbent assay for measurement of human immunoglobulin G antibodies to anthrax toxins. Clin Diagn Lab Immunol 2004;11:50-5.
- Khan SS, Smith MS, Reda D, Suffredini AF, McCoy JP. Multiplex bead array assays for detection of soluble cytokines: Comparisons of sensitivity and quantitative values among kits from multiple manufacturers. Cytometry 2004;61:35-9.
- Jani IV, Janossy G, Brown DW, Mandy F. Multiplexed immunoassays by flow cytometry for diagnosis and surveillance of infectious diseases in resource-poor settings. Lancet Infect Dis 2002;2:243-50.
- Faucher S, Martel A, Sherring A, Ding T, Malloch L, Kim JE, et al. Protein bead array for the detection of HIV-1 antibodies from fresh plasma and dried-blood-spot specimens. Clin Chem 2004;50:1250-3.
- Bellisario R, Colinas RJ, Pass KA. Simultaneous measurement of thyroxine and thyrotropin from newborn dried blood-spot specimens using a multiplexed fluorescent microsphere immunoassay. Clin Chem 2000;46:1422-4.
- Lukacs Z, Mordac C, Kohlschutter A, Kruithof R. Use of microsphere immunoassay for simplified multianalyte screening of thyrotropin and thyroxine in dried blood spots from newborns. Clin Chem 2003;49:335-6.
- Kellar KL, Iannone MA. Multiplexed microsphere-based flow cytometric assays. Exp Hematol 2002;30:1227-37.
- Carson RT, Vignali DA. Simultaneous quantitation of 15 cytokines using a multiplexed flow cytometric assay. J Immunol Methods 1999;227:41-52.
- Wong SJ, Demarest VL, Boyle RH, Wang T, Ledizet M, Kar K, et al. Detection of human anti-flavivirus antibodies with a West Nile Virus recombinant antigen microsphere immunoassay. J Clin Microbiol 2004;42:65-72.
- Kellar KL, Douglass JP. Multiplexed microsphere-based flow cytometric immunoassays for human cytokines. J Immunol Methods 2003;279:277-85.
- Pataki J, Szabo M, Lantos E, Szekvolgyi L, Molnar M, Hegedus E, et al. Biological microbeads for flow-cytometric immunoassays, enzyme titrations, and quantitative PCR. Cytometry A 2005;68:45-52.