
Induction of leukemia cell apoptosis by cheliensisin A involves down-regulation of Bcl-2 expression1
Introduction
Apoptosis is defined by distinct morphological and biochemical changes, mediated by a family of cysteine aspases (caspases) that are expressed as inactive zymogens and are proteolytically processed to an active state following an apoptotic stimulus[1]. Two pathways leading to caspase cascade activation, including an intrinsic pathway and an extrinsic pathway, have been characterized[2,3]. Both pathways may involve the release of mitochondrial proteins, such as cytochrome c, Smac/DIABLO, and HtRA2, into the cytosol to trigger the activation of caspases, including initiator caspase-9 and effector caspase-3. The release of mitochondrial proteins into the cytosol usually depends on mitochondrial membrane permeabilization, which is regulated by pro- and anti-apoptotic Bcl-2 family members[1]. In theory, factors capable of regulating the activities of caspases and/or the functions of Bcl-2 family members are able to induce or prevent apoptotic cell death.
Styryl-lactones are a new class of compounds with potential anti-tumor activities[4,5]. Most styryl-lactones are isolated from the genus Goniothalamus (Annonaceae)[4]. The mechanisms of action by which styryl-lactones exert their anti-tumor activity are currently unknown. The suggested mechanisms include non-steroid, receptor-mediated anti-proliferative effects[6], disruption of mitochondrial transmembrane potential[7], and the induction of apoptosis[8]. GC-51, a novel styryl-lactone isolated from Goniothalamus cheliensis, has previously been shown in our laboratory to possess potent cytotoxicity against human promyelocytic leukemia HL-60 cells[9]. In the present study, we investigated the potential apoptosis-inducing effect of GC-51 and the possible mechanism of action involved.
Materials and methods
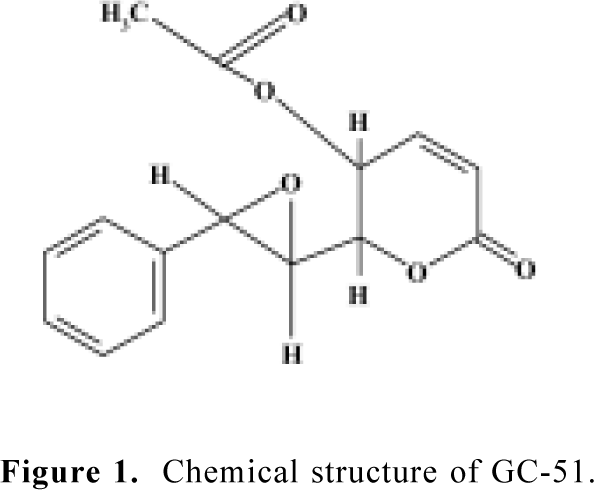
Chemicals GC-51 [6(7,8-epoxy-styryl)-5-acetoxy-5,6-dihydro-2-pyrone] was purified from Goniothalamus cheliensis at the Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China, as previously described[9] (Figure 1). GC-51 is a white crystal powder and its purity is greater than 99.0%. H-89 and chelerythrine chloride were purchased from Calbiochem (San Diego, USA). Propidium iodide and 4',6'-diamino-2-phenylindole (DAPI) were purchased from Sigma (St Louis, USA). RNase, polyclonal antibodies against caspase-3 and PARP were purchased from Becton Dickinson (Franklin Lakes, USA). Trizol and RPMI-1640 medium were purchased from Gibco (Grand Island, New York, USA).
Cell culture and treatment Human promyelocytic leukemia HL-60 cells were from the American Type Culture Collection (Manassas, VA 20108 USA). HL-60 cells were maintained in RPMI-1640 medium containing 10% heat-inactivated fetal bovine serum, 100 kU/L penicillin, and 100 mg/L streptomycin. Cells were cultured in a humidified incubator at 37 °C in 5% CO2/air. GC-51 was dissolved in dimethylsulfoxide and diluted in culture medium just prior to use. The final concentration of dimethylsulfoxide was less than 0.1% which had no effect on cell proliferation and the assay system.
Morphology staining Morphological changes in HL-60 cells were determined by staining the cells with DNA-specific dye, DAPI, after the GC-51-treated cells were fixed with methanol for 10 min at room temperature. Stained cells were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
Cell cycle analysis The DNA content of the cells was analyzed as previously described[10,11]. In brief, untreated and treated cells were harvested by centrifugation, washed in phosphate-buffered saline (PBS containing 137 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, 1.8 mmol/L KH2PO4, pH 7.4), and fixed in ice-cold 70% ethanol overnight. Following fixation, the cells was stained with PBS containing 50 mg/L propidium iodide, 0.1% Triton X-100, and 20 mg/L RNase. The fluorescence of individual cells was measured using FACStar plus flow cytometer (Becton Dickinson, Franklin Lakes, USA).
DNA agarose gel electrophoresis Analysis of DNA fragmentation was carried out using DNA agarose gel electrophoresis as described previously[11]. In brief, DNA extracted from untreated and treated cells was loaded onto a 1.8% agarose gel in TBE (Tris 45 mmol/L borate buffer, edetic acid 1 mmol/L, pH 8.0), and electrophoresed at 40 V for 5 h. DNA in the gels was visualized under UV light after staining with ethidium bromide 5 mg/L.
Western blotting Western blot analysis was carried out as described previously[12,13]. After treatment with GC-51, cells were washed twice with ice-cold PBS and total cell lysates were collected in sodium dodecylsulfate (SDS) sample buffer (50 mmol/L Tris-HCl, pH 6.8, 100 mmol/L DTT, 2% SDS, 0.1% bromophenol blue, 10% glycerol). Cell lysates containing equal amounts of protein were separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidine difluoride membranes. After blocking in 5% non-fat milk in TBST (Tris-buffered saline with 0.1% Tween 20, pH 8.0), the membranes were incubated with the appropriate primary antibodies at 4°C overnight. Membranes were washed three times in TBST and exposed to secondary antibodies for 2 h at room temperature, and then washed four times in TBST. Immunoreactive proteins were visualized using the enhanced chemiluminescence system from Pierce (Rockford, IL, USA).
RNA isolation and RT-PCR Total mRNA was prepared from HL-60 cells using Trizol (Sangon, Shanghai, China). The cDNA was synthesized using random hexamers from 1 μg of mRNA. To amplify the cDNAs, a 2 μL aliquot of the reverse-transcribed cDNA was subject to 29 cycles of PCR in 50 μL of 1×buffer (10 mmol/L Tris·HCl, pH 8.3, 1.5 mmol/L MgCl2, 50 mmol/L KCl, 50 μmol/L dNTP, three units of Taq DNA polymerase and 0.2 μmol/L specific primers). Each cycle consisted of denaturation at 94 °C for 30 s, annealing at 60 °C for 40 s, and extension at 72 °C for 40 s. The amplified products were separated by electrophoresis on 1.0% agarose gel. Each RT-PCR was repeated three times using different preparations of RNA. The RT-PCR of GAPDH was used as an internal control with all samples. The PCR primers used are listed in Table 1.

Full table
Statistical analysis Data were analyzed using Student’s t-tests. P values less than 0.05 were considered significant.
Results
Inhibition of cell proliferation and induction of cell apoptosis by GC-51 The effect of GC-51 on cell proliferation was determined by counting the cell number using the Trypan blue exclusion method. After a 48-h treatment, GC-51 markedly inhibited the proliferation of HL-60 cells in a concentration-dependent manner with an IC50 of 2.4±0.2 μmol/L (95% confidence: 1.5–3.3 μmol/L, Figure 2A). The inhibition of cell proliferation was also time-dependent (data not shown).
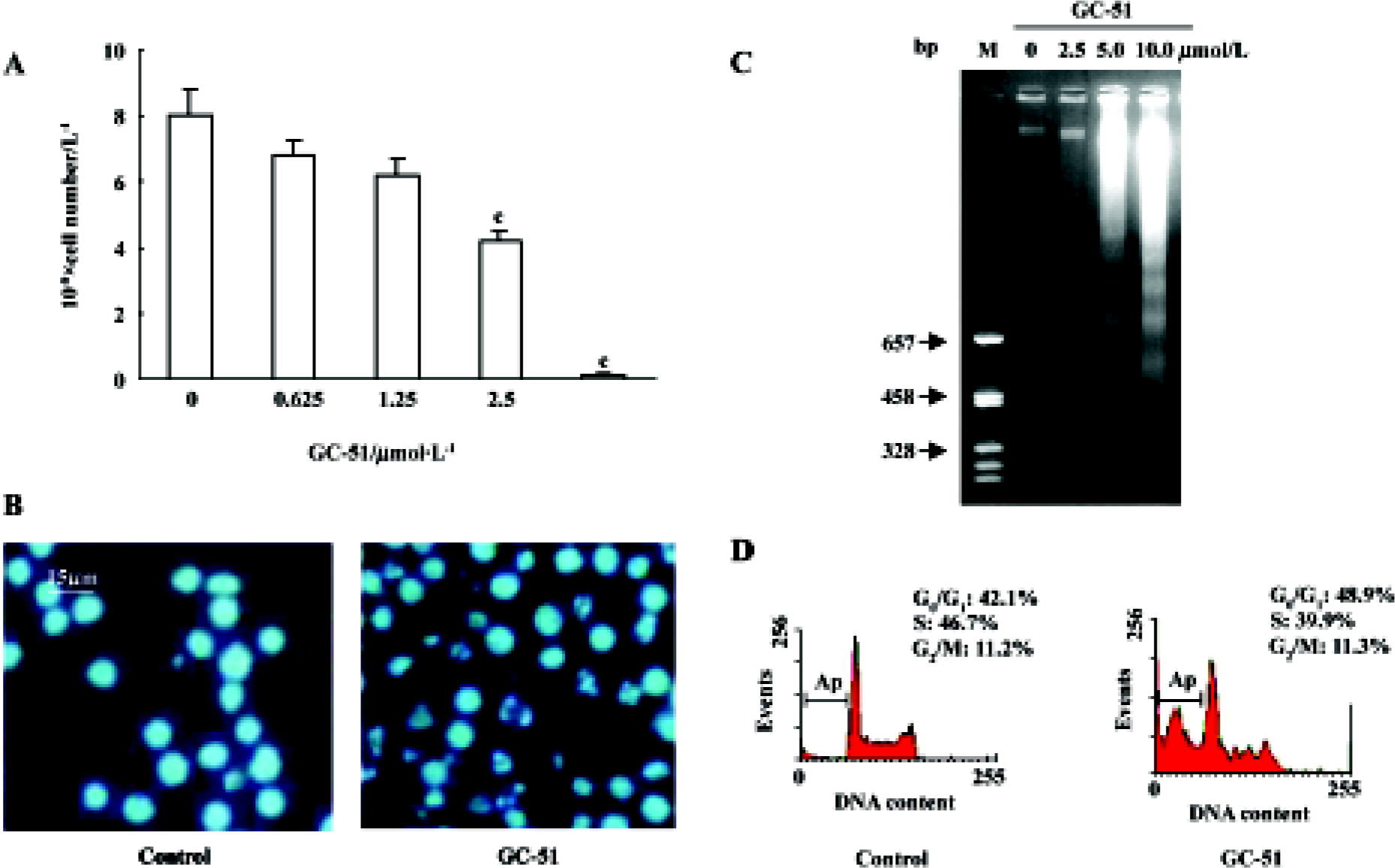
To further identify the inhibitory effect of GC-51 on cell proliferation, we examined the apoptosis-inducing effect of GC-51 on HL-60 cells. Incubation of HL-60 cells with GC-51 (2.5–10 μmol/L) for various times (2–8 h) resulted in dramatic morphological changes in the treated cells typical of apopto-sis, such as cell shrinkage, chromatin condensation, and nuclear fragmentation (Figure 2B). Treatment of cells with 10 μmol/L GC-51 for 4 h was enough to induce cell apoptosis. The apoptosis-inducing effect of GC-51 was confirmed by the appearance of a DNA “ladder”, another major hallmark of apoptosis (Figure 2C).
It is widely accepted that apoptotic cells have reduced DNA stainability following staining with a variety of fluorochromes, including propidium iodide. Thus, the appearance of cells with low stainability (ie sub-G1 peak) in cultures has been considered to be another marker of apoptosis and has been used to quantify the extent of apoptosis. GC-51 treatment led to the formation of a “sub-G1” peak in the DNA content frequency distribution (Figure 2D) and caused apoptosis in a concentration- and time-dependent manner. Exposure of HL-60 cells to 10 μmol/L GC-51 for 2 h, 6 h, and 8 h resulted in 3.5%±3.0%, 26.2%±4.3%, and 53.0%±7.2% cells undergoing apoptosis, respectively, whereas GC-51 treatment for 8 h at concentrations of 5, 10, and 25 μmol/L led to apoptotic cell death in 7.3%±3.5%, 54.2% ±9.2%, and 78.7%±10.2% cells, respectively.
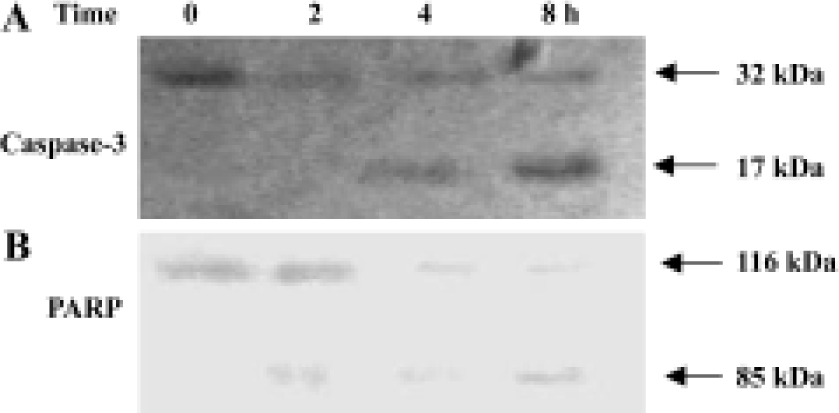
Activation of caspase-3 in GC-51-treated apoptotic cells Cell death is executed by effector caspases such as caspases-3, thus, the activation of caspase-3 is the critical cellular event during apoptosis. Intact caspase-3 was a 32 kDa protein as detected in control cells and was processed into its catalytically active p17 subunits in GC-51-treated HL-60 cells (Figure 3A). To confirm the activation of caspase-3, we evaluated the cleavage of PARP, a major substrate of caspase-3. GC-51 treatment caused the cleavage of 116 kDa PARP into a 85 kDa form (Figure 3B), indicating that caspase-3 was indeed activated during GC-51-induced HL-60 cell apoptosis.
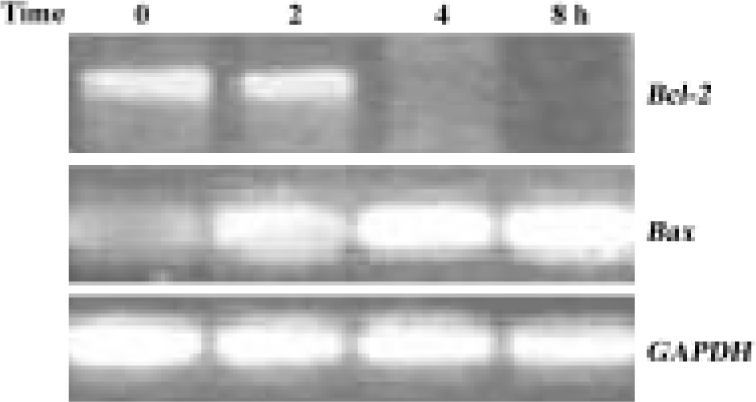
Downregulation of Bcl-2 mRNA expression by GC-51 Caspase-3 activation requires the release of a number of proteins, including cytochrome c, from mitochondria and this release is, in general, determined by the permeability of the mitochondrial membrane. Members of the Bcl-2 family regulate the release of cytochrome c from mitochondria, primarily by affecting the permeability of the mitochondrial membrane. Thus, we tested the effect of GC-51 on the expression of the anti-apoptotic gene Bcl-2 and the pro-apoptotic gene Bax. Exposure of HL-60 cells to 10 μmol/L GC-51 for 8 h led to a significant reduction in Bcl-2 gene expression. In contrast, the expression of the Bax gene markedly increased after GC-51 treatment. As a control, GC-51 had no effect on the expression of the GAPDH gene (Figure 4). Regulation by GC-51 of the expression of the Bcl-2 and Bax genes resulted in a decrease in the ratio of Bcl-2/Bax, which will make the cells more susceptible to apoptosis inducers.
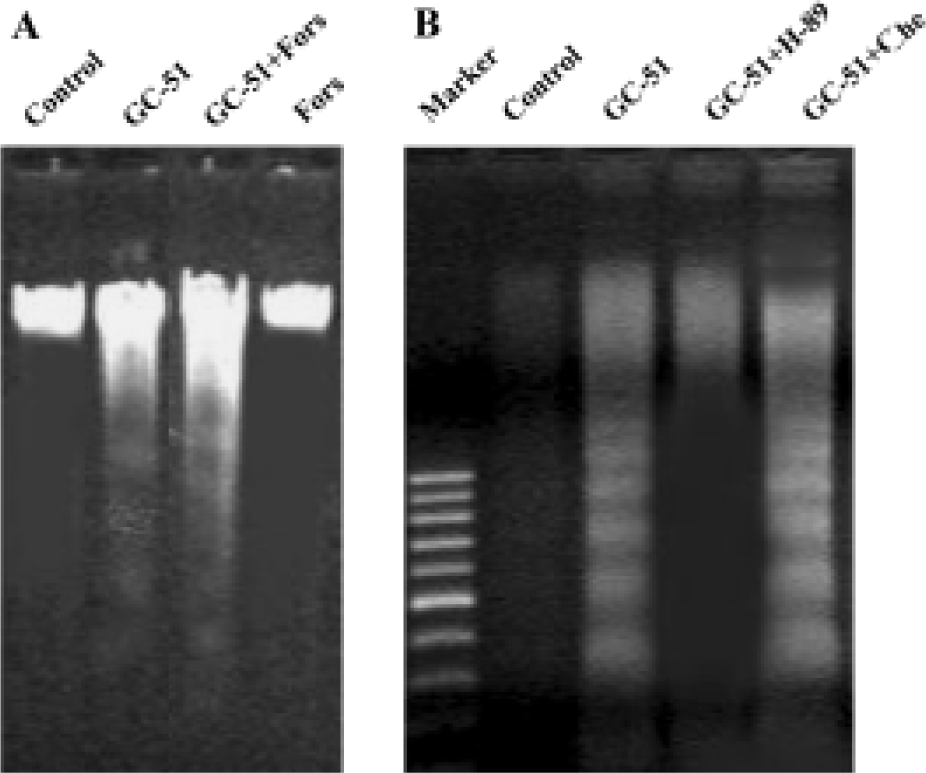
Induction of apoptosis by GC-51 in HL-60 cells was PKA dependent Bullatacin, a bioactive component isolated from Annonaceous, induces cell apoptosis by reducing the intracellular cAMP level[14]. Thus, we tested the role of cAMP in GC-51-induced HL-60 cell apoptosis. If induction of HL-60 cell apoptosis by GC-51 occurs via a similar mechanism to that of bullatacin, forskolin, an adenylate cyclase activator that effectively increases intracellular cAMP level, should block the apoptosis-inducing effect of GC-51. We did not record a significant effect of forskolin on induction of apoptosis by GC-51 in HL-60 cells (Figure 5A). To our surprise, H89, a PKA-specific inhibitor, blocked induction of HL-60 apoptosis by GC-51, whereas chelerythrine, a protein kinase C inhibitor, had no effect on the action of GC-51 (Figure 5B), suggesting that GC-51 induction of HL-60 apoptosis is specifically dependent on PKA activity.
Discussion
Defects in apoptosis underpin both tumorigenesis and drug resistance, and most anticancer drugs exert their chemotherapeutic effect by inducing tumor cell apoptosis[1]. Therefore, the discovery and development of drugs targeting the apoptotic pathway represent a novel and rational strategy for the treatment of cancers.
GC-51 a novel styryl-lactone isolated from Goniothalamus cheliensis, which is a widely occurring plant in Southwest China, exhibits a potent cytotoxicity in HL-60 cells[9]. In the present study, GC-51 significantly inhibited HL-60 cell proliferation, which is consistent with a previous report[9]. GC-51 inhibits cell proliferation or induces cell apoptosis based on the concentrations applied (low vs high concentra-tions). Induction of apoptosis by GC-51 was confirmed by the appearance of characteristic morphological changes, including a DNA “ladder” and a “sub-G1 peak” in DNA content frequency distribution. Our results demonstrate that GC-51, like other styryl-lactones, is a potent inducer of cell apoptosis.
Caspase-3 is an effector caspase whose activation is critical for the execution of apoptotic death of tumor cells. We clearly showed in the present study that caspase-3 was activated during GC-51-induced HL-60 cell apoptosis and the activation was further verified by the cleavage of PARP, a preferred substrate of caspase-3 in apoptotic cells. Our results combined with findings from other laboratories[7,8] suggest that the activation of caspase-3 is a common mechanism responsible for styryl-lactone-induced apoptosis.
It is generally accepted that the ratio of Bcl-2/Bax determines the permeability of mitochondrial transmembranes, and this permeability controls the release of a number of proteins, such as cytochrome c, from the mitochondria into the cytosol[1]. GC-51 treatment significantly inhibited the expression of anti-apoptotic gene Bcl-2 and increased the expression of the pro-apoptotic gene Bax. This effect of GC-51 on the expression of members of the Bcl-2 family is, at least in part, contributing to the activation of caspase-3 and the subsequent apoptosis of HL-60 cells. To our knowledge, this is the first study to show that GC-51, a styryl-lactone compound, induces HL-60 cell apoptosis via reduction of the expression of the Bcl-2 gene.
cAMP is a critical mediator of apoptosis[15-18]. cAMP triggers apoptosis by stimulating its major effector protein PKA, which phosphorylates the cAMP response element-binding protein (CREB), activates caspase-3, and induces apoptosis[18]. Kim et al[19] showed that 8-Cl-cAMP, an analogue of cAMP, induced cell-cycle-specific apoptosis in HL-60 cells. In contrast, PKA phosphorylates Bad, a member of the Bcl-2 family, to prevent apoptosis[1]. Bullatacin induces hepatoma cell apoptosis by decreasing the intracellular cAMP level[14]. In the present study, we examined whether GC-51 induced HL-60 cell apoptosis via a similar mechanism. The data showed that forskolin had no significant effect on induction of cell apoptosis by GC-51, whereas in contrast H-89, the PKA-specific inhibitor, blocked the apoptosis-inducing effect of GC-51. Our results suggest that GC-51 induction of apoptosis is dependent on PKA. The opposite effects of GC-51 and bullatacin on intracellular cAMP levels is not surprising considering that these two compounds are totally different in structure: GC-51 is a styryl-lactone compound, whereas bullatacin is an annonaceous acetogenin compound. In addition, the cells used in these two experiments were also different, leukemia HL-60 cells compared with hepatoma 2.2.15 cells, and it is well known that effect of cAMP on cell apoptosis is cell-type specific[18]. We postulate that GC-51 induces HL-60 cell apoptosis via a similar mechanism to that demonstrated by Zhang and Insel[18]. However, further investigations are needed to confirm this hypothesis.
In conclusion, GC-51 effectively induces HL-60 apoptosis. The apoptosis-inducing effect of GC-51 is PKA-dependent and involves the downregulation of anti-apoptotic Bcl-2 gene expression and the activation of caspase-3.
References
- Johnstone R, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002;108:153-64.
- Green DR. Apoptotic pathways: paper wraps stone blunts scissors. Cell 2000;102:1-4.
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001;15:2922-33.
- Mereyala HB, Joe M. Cytotoxic activity of styryl lactones and their derivatives. Curr Med Chem Anti-Canc Agents 2001;1:293-300.
- Mu Q, Tang WD, Liu RY, Li CM, Lou LG, Sun HD, et al. Constituents from the stems of Goniothalamus griffithii. Planta Med 2003;69:826-30.
- Pihie AH, Stanslas J, Din LB. Non-steroid receptor-mediated antiproliferative activity of styrylpyrone derivative in human breast cancer cell lines. Anticancer Res 1998;18:1739-43.
- Inayat-Hussain SH, Annuar BO, Din LB, Ali AM, Ross D. Loss of mitochondrial transmembrane potential and caspase-9 activation during apoptosis induced by the novel styryl-lactone goniothalamin in HL-60 leukemia cells. Toxicol 2003;17:433-9.
- Inayat-Hussain SH, Osman AB, Din LB, Ali AM, Snowden RT, MacFarlane M, et al. Caspases-3 and -7 are activated in gonio-thalamin-induced apoptosis in human Jurkat T-cells. FEBS Lett 1999;456:379-83.
- Li CM, Mu Q, Sun HD, Xu B, Tang WD, Zheng HL, et al. A new anti-cancer constituent of Goniothalamus cheliensis. Acta Bot Yunnan 1998;20:102-4.
- Lou LG, Xu B. Induction of apoptosis of human leukemia cells by α-anordrin. Chin J Cancer Res 1997;9:1-5.
- Lou LG, Xu B. Induction of apoptosis in human leukemia K562 cells by α-anordrin. Acta Pharmacol Sin 1996;17:255-8.
- Lou LG, Urbani J, Riberio-Neto F, Altschuler DL. cAMP inhibition of Akt is mediated by activated and phosphorylated Rap1b. J Biol Chem 2002;277:32799-806.
- Ribeiro-Neto F, Urbani J, Lemee N, Lou L, Altschuler DL. On the mitogenic properties of Rap1b: cAMP-induced G1/S entry requires activated and phosphorylated Rap1b. Proc Natl Acad Sci USA 2002;99:5418-23.
- Chiu HF, Chih TT, Hsian YM, Tseng CH, Wu MJ, Wu YC. Bullatacin, a potent antitumor Annonaceous acetogenin, induces apoptosis through a reduction of intracellular cAMP and cGMP levels in human hepatoma 2.2.15 cells. Biochem Pharmacol 2003;65:319-27.
- Muhl H, Nitsch D, Sandau K, Brune B, Varga Z, Pfeilschifer J. Apoptosis is triggered by cyclic AMP signaling pathway in renal mesangial cells. FEBS Lett 1996;382:271-5.
- Lomo J, Blomhoff HK, Beiske K, Stokke T, Smeland EB. TGF-β1 and cyclic AMP promote apoptosis in resting human B lymphocytes. J Immunol 1995;154:1634-43.
- Aharoni D, Dantes A, Oren M, Amsterdam A. cAMP-mediated signals as determinants for apoptosis in primary granulosa cells. Exp Cell Res 1995;218:271-82.
- Zhang L, Insel PA. The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A-and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem 2004;279:20858-65.
- Kim SN, Ahn YH, Kim SG, Park SD, Cho-Chung YS, Hong SH. 8-Cl-cAMP induces cell cycle-specific apoptosis in human cancer cells. Int J Cancer 2001;93:33-41.