
Design, synthesis, and anti-inflammatory evaluation of a series of novel amino acid-binding 1,5-diarylpyrazole derivatives
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used in the treatment of pain and inflammation. NSAIDs act by inhibiting the catalytic activity of cyclooxy-genase (COX), which results in a blockage of the formation of prostaglandins (PGs). COX exists as two distant isoforms (COX-1 and COX-2)[1,2]. Inhibition of COX-2 accounts for the anti-inflammatory effects of NSAIDs, whereas interruption of COX-1 leads to gastrointestinal toxicity[2,3]. Therefore, it is proposed that a selective COX-2 inhibitor would have a superior safety profile[4] to traditional NSAID.
Since the discovery of COX-2, a large number of selective COX-2 inhibitors have been described[5–8], of which vicinal diaryl heterocycles represent the most important group[9–12]. These selective COX-2 inhibitors have pronounced anti-inflammatory effects with reduced or no gastrointestinal side effects. Celecoxib, in the 1,5-diarylpyrazole class of compound[13], was the first launched selective COX-2 inhibitor, and has excellent selectivity and potent anti-inflammatory activity; however, its aqueous solubility is relatively low, which decreases its oral bioavailability[14]. One approach to address this problem is to convert the compound into a prodrug that is readily soluble in water. Researchers attempted to create a water-soluble form by N-acetylation of the sulfonamide group of celecoxib followed by preparation of its sodium salt[15], but the result was unsatisfactory. The acetylation product had a lower level of inhibitory effect on rat paw edema than that produced by celecoxib.
Recently, the cardiovascular safety of coxibs has raised serious concerns; Merck & Co even voluntarily withdrew a rofecoxib product from the market because of an increased risk of cardiovascular events. Although there has been concern about adverse cardiovascular events associated with the use of selective COX-2 inhibitors[16], there is still no evidence indicating that cardiac toxicity is linked with all COX-2 inhibitors[17,18]. Selective COX-2 inhibitors are still under development[5,19,20]. It has been proposed that drugs with higher selectivity for COX-2 tend to induce cardiovascular disease[18,21]. Therefore, we chose the moderately selective COX-2 inhibitor, celecoxib, as a target for modification (the COX-1/COX-2 IC50 ratios of etoricoxib, rofecoxib, valdecoxib and celecoxib are 106, 35, 30, and 7.6, respectively).
To improve the oral absorption of celecoxib, a prodrug strategy was used in this study. In our experiments, we were mindful of the following: (1) conserving the main structure of celecoxib, including the trifluoromethyl and benzenesul-fonamide groups, which contribute to its inhibitive qualities and selectivity of COX-2[13]; (2) partially containing the methyl group in cycle C, because it can be metabolized to the corresponding hydroxymethyl and carboxylic acid analogs (glucuronide conjugation of the carboxylic acid metabolite is a major pathway of elimination in humans[22]); (3) introducing some polar groups, such as amino or methylamino groups, into cycle C; (4) binding with a natural amino acid followed by preparation of its hydrochloride acid salt. The amide bonds are inclined to be hydrolyzed in vivo, and the cleaved natural amino acid part is safe for humans.
Based on these considerations, in the present study, we designed and synthesized some 1,5-diarylpyrazole derivatives. These hydrochloride salts were expected to have good aqueous solubility and act like prodrugs, which can exhibit potent anti-inflammatory activity in vivo after being converted to their parent compounds.
Materials and methods
Animals Female Kun-ming mice (grade SPF II, certificate N
Chemicals and reagents Substituted acetophenone and ethyl trifluoracetate were purchased from Aldrich (St Louis, MO, USA) and Acros (Geel, Belgium). The tert-butoxycarbonyl (BOC)-protected amino acid was from GL Biochem (Shanghai, China). The other reagents were from the Shanghai Chemical Reagent Co (Shanghai, China). All the chemicals were analytical grade or above.
Chemistry To obtain the expected prodrugs, celecoxib (positive drug), and celecoxib analogs (parent compounds) containing amino or methylamino groups were synthesized first as follows.
4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (celecoxib) Celecoxib (com-pound 2) was synthesized as described previously[13] with some modifications. In first step, sodium methanol was replaced with sodium ethanol, and the reaction time was reduced from 24 h to 4 h. In second step, dione 1 reacted with (4-sulfamoylphenyl)hydrazine hydrochloride (self-preparation) to produce celecoxib in the form of white crystals (Scheme 1, a-b). Purity as assessed by high performance liquid chromatography (HPLC) was 99.8%. The structure was identified by EI-MS, Mp, and H1NMR.
Celecoxib analogs containing amino or methylamino groups (parent compounds) Three classes of 6 compounds were synthesized as followed.
All the 6 compounds were identified by EI-MS, Mp, and H1NMR, and their purities as determined by HPLC were all above 99.3%.Synthesis of prodrug derivatives The 3 classes of cele-coxib analogs containing amino or methylamino groups reacted with several kinds of tert-BOC-protected amino acids in the presence of 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride (EDCI). Then the intermediates bound with the BOC group were deprotected with saturated hydrochloride-ethyl acetate solution to obtain the final amino acid hydrochloride salt (Scheme 3). All products were checked by EI-MS, Mp, H1NMR, IR, and HRMS.
Biological assays
In vitro biochemical assays Cell-based assays were performed as described previously[25]. In brief, the drugs were pre-incubated with infected Spodoptera frugiperda (Sf9) cells for 15 min prior to a 10 min challenge with 10 μmol/L arachidonic acid. The concentration of prostaglandin E2 (PGE2) was determined by interpolation from a standard curve and inhibition was calculated by comparison of PGE2 production by drug-treated cells with that of Me2SO-treated cells.
In vivo screening methods. Carrageenan-induced rat paw edema[26] Test compounds were suspended in 0.5% carboxymethylcellulose solution and intragastrically administered to rats at three doses (25 mg/kg, 50 mg/kg and 100 mg/kg) before carrageenan was injected. The control group rats received the same volume of vehicle (0.5% carboxymethylcellulose solution) according to their weight. Paw edema was then induced by subplantar injection of 50 µL 1% (w/v) sterile carrageenan in saline into the right hindpaw. Paw volume was measured before and 3 h after carrageenan injection with a plethysmometer (Shandong Academy of Medical Sciences, China). The extent of paw edema in the treatment group was compared to that of the vehicle control group and percentage inhibition was calculated in comparison to the vehicle group.
In the preliminary screening of mice in vivo, the inflammatory index was paw weight instead of volume, which was used for the rats.
Preliminary pharmacokinetic study of prodrugs As described previously[27], HPLC/MS was used to confirm the conversion of prodrug-like derivatives in vivo in rats. A single dose (100 mg/kg) of the selected compound (that showed potent inhibitory effects on rat paw edema), was orally administered to rats (n=6). Blood samples (50 µL) were taken before and 0.5 h, 2 h, 6 h, 12 h, 18 h, 24 h, 30 h, 36 h, 48 h, 60 h, and 72 h after intragastric administration, and immediately centrifuged at 10 000×g at 4 °C for 15 min. The plasma samples were stored at -20 °C.
Plasma concentration-time curves were evaluated by InnaPhase Kinetica (InnaPhase Corporation, Philadelphia, PA, USA)
Statistical analysis Data were expressed as mean±SEM (unless noted otherwise), and subjected to one-way analysis of variance (ANOVA) and Dunnett’s test. P<0.05 was considered to be statistically significant.
Results
Chemistry Celecoxib, 6 of the 1,5-diarylpyrazole class of celecoxib analogs, and their amino acid derivatives (hydrochloride salts) were synthesized. Three kinds of BOC-protected natural amino acids were selected for introduction. They were glycine, L-alanine, and L-phenylalanine. The main analytical data, from 1H NMR and HRMS, are summarized in Table 1.
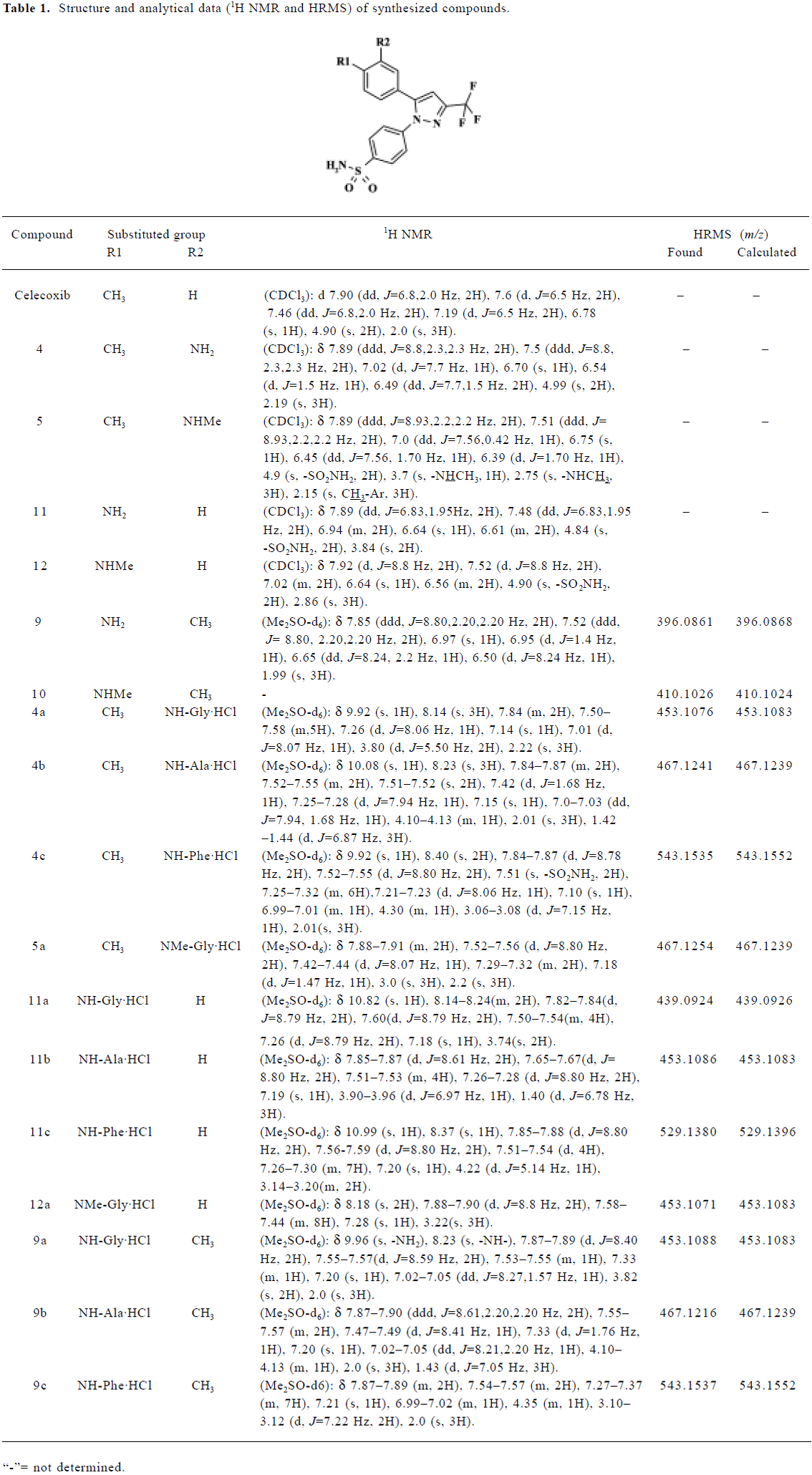
Full table
The aqueous solubility of the hydrochloride salts was greatly improved compared with that of their parent compounds and celecoxib. Their aqueous solubilities were, respectively, 10–33 g/L, 0.1–1 g/L, and <0.05 g/L at 18 °C.
In vitro biochemical screening All the compounds were evaluated using the intact cell assay. Parent compounds 4, 5, 9, and 10 exhibited potent and selective inhibition of COX-2 activity in vitro (Table 2), whereas the inhibitory activities of the hydrochloride salts were greatly decreased compared with their parent compounds.
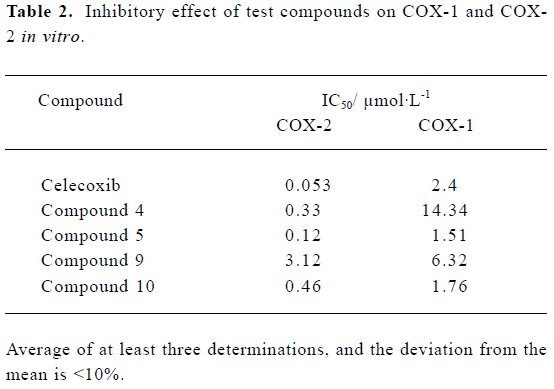
Full table
In vivo biological evaluation Although none of the hydrochloride salts had any anti-inflammatory activity in vitro, they could be expected to exert anti-inflammatory effects once they were converted into their parent compounds.
Based on our preliminary biological evaluation in mice (data not shown), compound 4a was selected for further evaluation in carrageenan-induced rat paw edema because of its potent inhibitory effect on edema. Three doses (25 mg/kg, 50 mg/kg and 100 mg/kg) of each of celecoxib, compound 4, and compound 4a, respectively, were orally administered to rats. Compound 4a had a high level of efficiency, which was identical to that of the possible parent compound 4 and similar to that of celecoxib (Table 3). Furthermore, compound 4a dose-dependently inhibited the inflammatory response to carrageenan-induced edema.
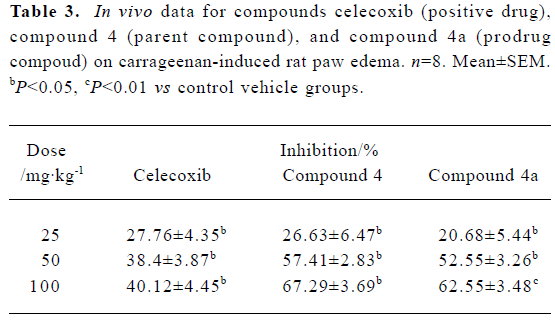
Full table
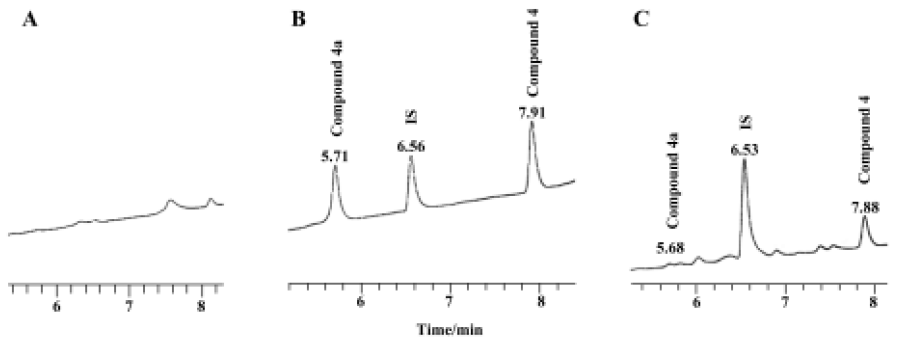
Preliminary pharmacokinetic study A preliminary pharmacokinetic study of compound 4a in rats (n=6) was undertaken to prove that it was converted in vivo to compound 4. Figure 1 shows the representative chromatograms of blank plasma, plasma containing compound 4, compound 4a, and internal standard (IS), and plasma sample added with IS 6 h after intragastric administration. The retention times of compound 4a, compound 4, and IS were 5.71 min, 7.91 min, and 6.56 min in blank plasma, respectively. In a plasma sample 6 h after intragastric administration, the retention times of compound 4a, compound 4 created from the conversion of compound 4a, and IS were 5.68 min, 7.88 min, and 6.53 min, respectively. The corresponding chromatography peak was identified by MS. We thus proved that compound 4a was actually converted to compound 4 after oral administration.
The pharmacokinetic parameters of compound 4 released from compound 4a were calculated by non-compartmental model analysis based on the drug concentration-time curve of 0–72 h sample dotting. Peak plasma concentration (Cmax), the corresponding time (Tmax), the area under the plasma concentration-time curve (AUC0-∞), and T1/2 were 7.7±1.1 mg/L, 13.2±2.7 h, 165.3±21.8 mg·L-1·h, and 6.4±1.3 h, respectively.
Discussion
In the present study, a series of amino acid-binding 1,5-diarylpyrazole derivatives were designed and synthesized as prodrugs. As expected, the aqueous solubility of these compounds was greatly improved compared with celecoxib. For example, the solubility of compound 4a in water at 18°C was 15–18 g/L, whereas that of celecoxib was <0.05 g/L. Good aqueous solubility should enhance the dissolution rate of a drug and thus improve its oral bioavailability[14].
In in vitro biological evaluations, it was found that the parent compounds 4, 5, 9, and 10 exhibited potent selectivity and inhibition of COX-2, although they all did not exceed celecoxib (Table 2). From the results, it was also found that the compounds containing the methylamino group had more potent inhibitory effects than those containing the amino group. In addition, all the hydrochloride salts showed extremely weak inhibition on COX activity.
Based on a previous hypothesis, the hydrochloride salt should bring about beneficial anti-inflammatory effects after delivering its parent compound in vivo. Therefore, anti-inflammatory evaluation in vivo was performed in carrageenan-induced mouse and rat paw edema models. Compound 4a brought about the expected potent inhibition of edema after intragastric administration. Furthermore, at doses of 50 mg/kg and 100 mg/kg, the anti-inflammatory effect of compound 4a was a little stronger than that of celecoxib, and identical to that of compound 4 (Table 3). Preliminary pharmacokinetic studies in rats also proved that compound 4a actually acted as a prodrug, which has an anti-inflammatory effect once it is converted into its active form, compound 4.
In the preliminary pharmacokinetic studies, the Tmax of compound 4 (as derived from compound 4a) was found at 12 h after oral administration; however, the Tmax of celecoxib was at 3 h after administration[28]. In our experiments, we only observed the inhibitory effects on edema at 3 h after oral administration. Therefore we predicted that when the concentration of compound 4 (as derived from compound 4a) reached a maximum, it should have stronger anti-inflammatory activity.
Although compound 4a did not exhibit more potency in vivo than its parent, compound 4, its potency was comparable with that of celecoxib (Table 3). We suppose that compound 4a was not completely converted to its parent compound, compound 4, or it produced other metabolisms in vivo. Similar prodrug conversion pharmacokinetics have been investigated by Mamidi et al[28]. Prodrugs derived from the same parent compound will have different activity in vivo when they are connected to different groups. Connecting more amino acids to the parent compound might offer a an exciting prospect for developing prodrug-like NSAIDs.
In summary, by introducing an amino or methylamino group, 6 celecoxib analogs were synthesized as parent compounds. The 6 compounds were combined with three kinds of natural amino acids to obtain a series of novel hydrochloride salts. By biological evaluation in vitro and in vivo, and by further preliminary pharmacokinetic studies, compound 4a was discovered, which was confirmed to act as a prodrug, and exhibited marked anti-inflammatory activity in vivo. This finding will be of great benefit to the structural modifications of prodrug-like selective COX-2 inhibitors.
References
- Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci USA 1991;88:2692-6.
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol 1998;38:97-120.
- Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem 1996;271:33157-60.
- Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA 1994;91:3228-32.
- Uddin MJ, Rao PN, McDonald R, Knaus EE. Design and synthesis of (E)-1,1,2-triarylethenes: novel inhibitors of the cyclo-oxygenase-2 (COX-2) isozyme. Bioorg Med Chem Lett 2005;15:439-42.
- Ranatunge RR, Augustyniak M, Bandarage UK, Earl RA, Ellis JL, Garvey DS, et al. Synthesis and selective cyclooxygenase-2 inhibitory activity of a series of novel, nitric oxide donor-containing pyrazoles. J Med Chem 2004;47:2180-93.
- Hinz B, Brune K. Cyclooxygenase-2: 10 years later. J Pharmacol Exp Ther 2002;300:367-75.
- Dannhardt G, Kiefer W. Cyclooxygenase inhibitors: current status and future prospects. Eur J Med Chem 2001;36:109-26.
- Chen XH, Bai JY, Shen F, Bai AP, Guo ZR, Cheng GF. Imrecoxib: a novel and selective cyclooxygenase 2 inhibitor with anti-inflammatory effect. Acta Pharmacol Sin 2004;25:927-31.
- Singh SK, Vobbalareddy S, Shivaramakrishna S, Krishnamraju A, Rajjak SA, Casturi SR, et al. Methanesulfonamide group at position-4 of the C-5-phenyl ring of 1,5-diarylpyrazole affords a potent class of cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem Lett 2004;14:1683-8.
- Ranatunge RR, Garvey DS, Janero DR, Letts LG, Martino AM, Murty MG, et al. Synthesis and selective cyclooxygenase-2 (COX-2) inhibitory activity of a series of novel bicyclic pyrazoles. Bioorg Med Chem 2004;12:1357-66.
- Uddin MJ, Rao PN, Knaus EE. Design and synthesis of novel celecoxib analogues as selective cyclooxygenase-2 (COX-2) inhibitors: replacement of the sulfonamide pharmacophore by a sulfonylazide bioisostere. Bioorg Med Chem 2003;11:5273-80.
- Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib). J Med Chem 1997;40:1347-65.
- Chawla G, Gupta P, Thilagavathi R, Chakraborti AK, Bansal AK. Characterization of solid-state forms of celecoxib. Eur J Pharm Sci 2003;20:305-17.
- Pal M, Madan M, Padakanti S, Pattabiraman VR, Kalleda S, Vanguri A, et al. Synthesis and cyclooxygenase-2 inhibiting property of 1,5-diarylpyrazoles with substituted benzenesulfonamide moiety as pharmacophore: preparation of sodium salt for injectable formulation. J Med Chem 2003;46:3975-84.
- Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med 2000;343:1520-8.
- Couzin J. Drug safety. Withdrawal of Vioxx casts a shadow over COX-2 inhibitors. Science 2004;306:384-5.
- Davies NM, Jamali F. COX-2 selective inhibitors cardiac toxicity: getting to the heart of the matter. J Pharm Pharm Sci 2004;7:332-6.
- Velazquez C, Rao PN, McDonald R, Knaus EE. Synthesis and biological evaluation of 3,4-diphenyl-1,2,5-oxadiazole-2-oxides and 3,4-diphenyl-1,2,5-oxadiazoles as potential hybrid COX-2 inhibitor/nitric oxide donor agents. Bioorg Med Chem 2005;13:2749-57.
- Chen QH, Rao PN, Knaus EE. Design, synthesis, and biological evaluation of N-acetyl-2-carboxybenzenesulfonamides: a novel class of cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem 2005;13:2459-68.
- Dogne JM, Supuran CT, Pratico D. Adverse cardiovascular effects of the coxibs. J Med Chem 2005;7:2251-7.
- Paulson SK, Vaughn MB, Jessen SM, Lawal Y, Gresk CJ, Yan B, et al. Pharmacokinetics of celecoxib after oral administration in dogs and humans: effect of food and site of absorption. J Pharma-col Exp Ther 2001;297:638-45.
- Benington F, Morin RD, Leiand CC. Mescaline analogs, VIII. Substituted 5-methoxy- and 5,6,7-trimethoxyindoles. J Org Chem 1958;23:19-23.
- Walker HG, Hauser CR. Synthesis of methyl ketones from diethyl acylmalonates. J Am Chem Soc 1946;68:1386-8.
- Zhang WY, Yang XN, Jin DZ, Zhu XZ, Chen XH, Bai JY, et al. Expression and enzyme activity determination of human cyclooxygenase-1 and -2 in a baculovirus-insect cell system. Acta Pharmacol Sin 2004;25:1000-6.
- Winter CA, Risley EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc Soc Exp Biol Med 1962;111:544-7.
- Werner U, Werner D, Pahl A, Mundkowski R, Gillich M, Brune K. Investigation of the pharmacokinetics of celecoxib by liquid chromatography-mass spectrometry. Biomed Chromatogr 2002;16:56-60.
- Mamidi RN, Mullangi R, Kota J, Bhamidipati R, Khan AA, Katneni K, et al. Pharmacological and pharmacokinetic evaluation of celecoxib prodrugs in rats. Biopharm Drug Dispos 2002;23:273-82.