
Remifentanil mimics cardioprotective effect of ischemic preconditioning via protein kinase C activation in open chest of rats
Introduction
Several studies found opioids preconditioning had a protective effect on the postischemic heart[1–5]. Remifentanil has been demonstrated to mimic the cardioprotective effect of ischemic preconditioning (IPC) in anesthetized open-chest rats, which reduced infarct size (IS) dose-dependently[5].
Remifentanil is an ultra-short-acting phenylpiperidine opioid analgesic agent, which is rapidly metabolized by nonspecific blood and tissue esterases[6]. It has an analgesic potency similar to fentanyl and 100 times greater than morphine[7]; the opioids that have been most extensively studied in cardioprotection. The effect of remifentanil preconditioning (RPC) was abolished by blockade of any one of the µ-, δ-, and κ-opioid receptors (OR). This means all three OR are involved in cardioprotection by RPC[5].
Opioid-induced cardioprotection and IPC seem to share a common pathway[8,9]. Several studies have demonstrated that fentanyl and morphine significantly reduced IS, and that this effect was blocked by the protein kinase C (PKC) inhibitor chelerythrine (CHE)[4,10]. However, no study has examined the role of PKC in the protection conferred by remifentanil-induced preconditioning.
Therefore, we decided to examine whether the protection effect of RPC on postischemic hearts was mediated by PKC in comparison with IPC.
Materials and methods
Surgical preparation Our preparation and measurements have been described previously in detail[5]. Briefly, male Sprague-Dawley rats weighing 300 to 350 g were used. The rats were anesthetized by intraperitoneal administration of pentobarbitone (50 mg/kg bodyweight) and maintained by repeat doses of 25 mg/kg every 60−90 min. All of the animals underwent tracheotomy and endotracheal intubation. Mechanical ventilation was provided with a Harvard Apparatus Rodent Respirator (Boston, MA, USA) and the rats were ventilated with room air at 60 to 70 breaths per min. Body temperature was monitored and maintained at 37±1 °C (mean±SD) using a heating pad. The carotid artery was cannulated to measure mean blood pressure (MBP) via a pressure transducer, and a Lead-II electrocardiogram, via subcutaneous stainless steel electrodes, monitored heart rate (HR). These were connected to a PowerLab monitoring system (ML750 PowerLab/4sp with MLT0380 Reusable BP Transducer, AD Instruments, USA). The right jugular vein was cannulated to infuse saline or drugs. A left thoracotomy was performed to expose the heart at the fifth intercostal space. After removing the pericardium, a 6-0 Prolene loop, along with a snare occluder, was placed at the origin of the left coronary artery (LCA). Regional ischemia was achieved by pulling the snare and securing the threads with a mosquito hemostat. Ischemia was confirmed by a substantial fall in left ventricular pressure, ECG changes, and cardiac cyanosis. After surgical preparation, the rat was allowed to stabilize for 15 min.
Study groups and experimental protocol Rats were randomly assigned to one of nine groups. All animals received 30 min of occlusion of the left coronary artery followed by 2-h reperfusion: Group 1, Control (CON, saline vehicle); Group 2, chelerythrine[11,12], (CHE, a PKC inhibitor, Sigma Chemical Co, Saint Louis, USA) 2 mg/kg iv 5 min before ischemia; Group 3, GF109203X[12], (GF, another potent and selective PKC inhibitor, Tocris Cookson Ltd, Bristol, UK) 0.05 mg/kg iv 5 min before ischemia; Group 4, RPC, RPC hearts were subject to three 5-min cycles of infusion of remifentanil (Glaxo Wellcome Operations, Greenford, Middlesex, UK) at 6 μg·kg-1·min-1 interspersed with 5-min drug-free periods before 30 min of occlusion of the left coronary artery and 2 h of reperfusion; Group 5, IPC, before the 30-min occlusion, rats were subjected to preconditioning by ischemia (IPC, 5-min occlusion, 5-min reperfusion×3); Group 6, CHE+RPC; Group 7, CHE+IPC (2 mg/kg, iv, 5 min before RPC or IPC); Group 8, GF +RPC; and Group 9, GF+IPC (0.05 mg/kg, iv, 5 min before RPC or IPC).
Determination of infarct size On completion of the reperfusion period, the heart was excised, transferred to a Langendorff apparatus, and perfused with normal saline for 1 min at a pressure of 100 cmH2O to flush out blood. The snare was securely re-tightened and 0.25% Evans blue dye was injected to stain the normally perfused region of the heart. This procedure allowed visualization of the normal, non-ischemic region and the area at risk (AAR). The heart was then weighed, frozen, and cut into 2-mm slices. Thereafter, the slices were stained by incubation at 37 °C for 20 min in 1% 2,3,5- triphenyltetrazolium (TTC, Sigma Chemical Co, Saint Louis, USA)[1,13,14] in phosphate buffer (pH 7.4), and then were immersed in 10% formalin, to enhance the contrast of the stain. The areas of infarct (TTC negative) and risk zone (TTC stained) for each slice were traced and digitized using a computerized planimetry technique (SigmaScan 4.0, Systat Software Inc, CA, USA). The volumes of the left ventricles, IS and AAR were calculated by multiplying each area with slice thickness and summing the product. The IS was expressed as a percentage of the AAR (IS/AAR).
Statistical analysis Data analysis was performed with a personal computer statistical software package (Prism v4.0, GraphPad Software, San Diego, USA). Data were expressed as mean±SD. Hemodynamics were analyzed using 2-way analysis of variance (ANOVA) with Bonferroni post-hoc test for multiple comparisons if significant F ratios were obtained. IS (expressed as percentage of the area at risk) were analyzed between groups using ANOVA with a Student-Newman-Keuls post-hoc test for multiple comparisons. Statistical differences were considered significant if P<0.05.
Results
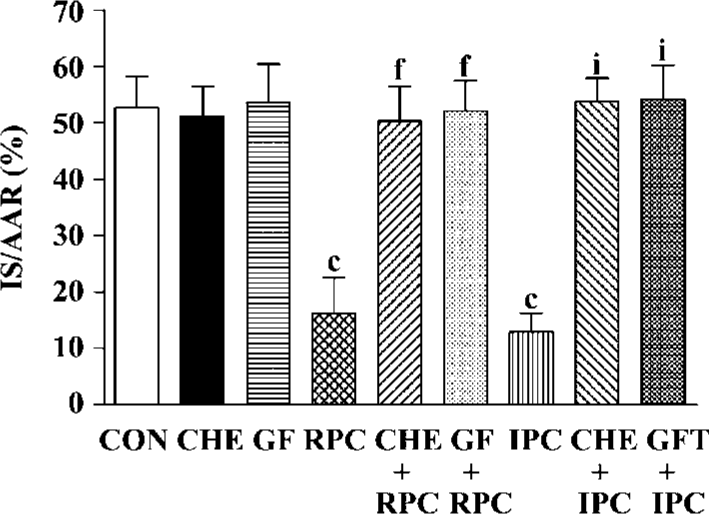
Effects of RPC and IPC on cardiac morphology LV+ RV volume average was 0.99±0.22 cm3 and the AAR ranged from 0.378±0.061 to 0.440±0.056 cm3. There was no difference in LV+RV and AAR between the control and treatment groups (Table 1). The IS, expressed as a percentage of the AAR, of the control group was 52.7%± 5.5%. In groups subjected to IPC and RPC the infarct sizes were significantly reduced. IPC and RPC markedly reduced IS/AAR from 52.7%±5.5% to 12.9%±3.4% (P<0.01 vs CON) and 16.2%±6.4% (P<0.01 vs CON), respectively. CHE 2 mg/kg, a PKC inhibitor, or GF 0.05 mg/kg, another selective PKC inhibitor, administered 5 min before RPC or IPC completely abolished the cardioprotective effect of RPC (IS/AAR: CHE+RPC 51.2%±5.0%, GF+RPC 53.6%±6.1%, P>0.05 vs CON) or IPC (CHE+IPC 53.7%±4.3%, GF+IPC 54.1%±6.2%, P>0.05 vs CON). Neither CHE nor GF by itself modified IS in non-PC hearts (Figure 1).
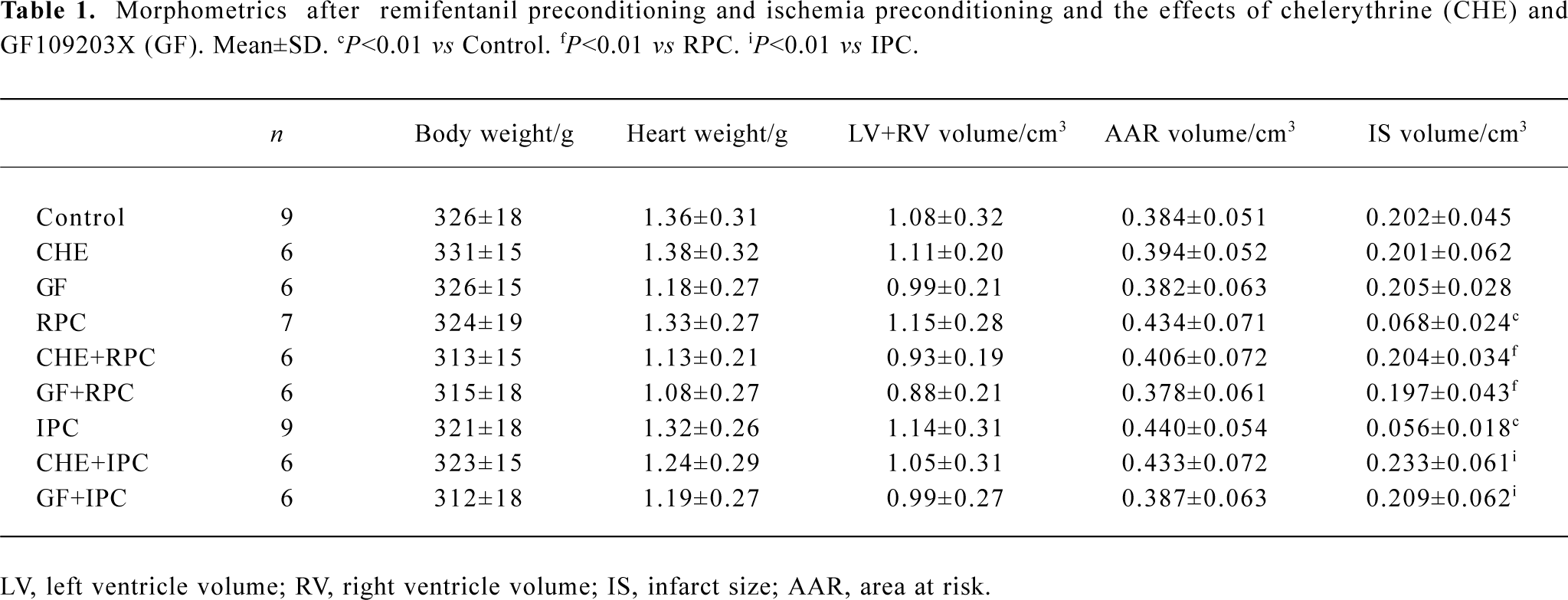
Full table
Effects of RPC and IPC on hemodynamics As shown in Table 2, administration of remifentanil at 6 μg·kg-1·min-1 significantly reduced the HR, MBP, and RPP (P<0.01 vs baseline). HR in CHE+RPC or GF+RPC group was not reduced significantly after pretreated with remifentanil, and the difference was significant compared with RPC group (P<0.05, respec-tively). The bradycardia produced by remifentanil was abolished with pretreatment of CHE or RPC. There was no difference in any of the hemodynamic parameters between control and treatment groups during ischemia and reperfusion.
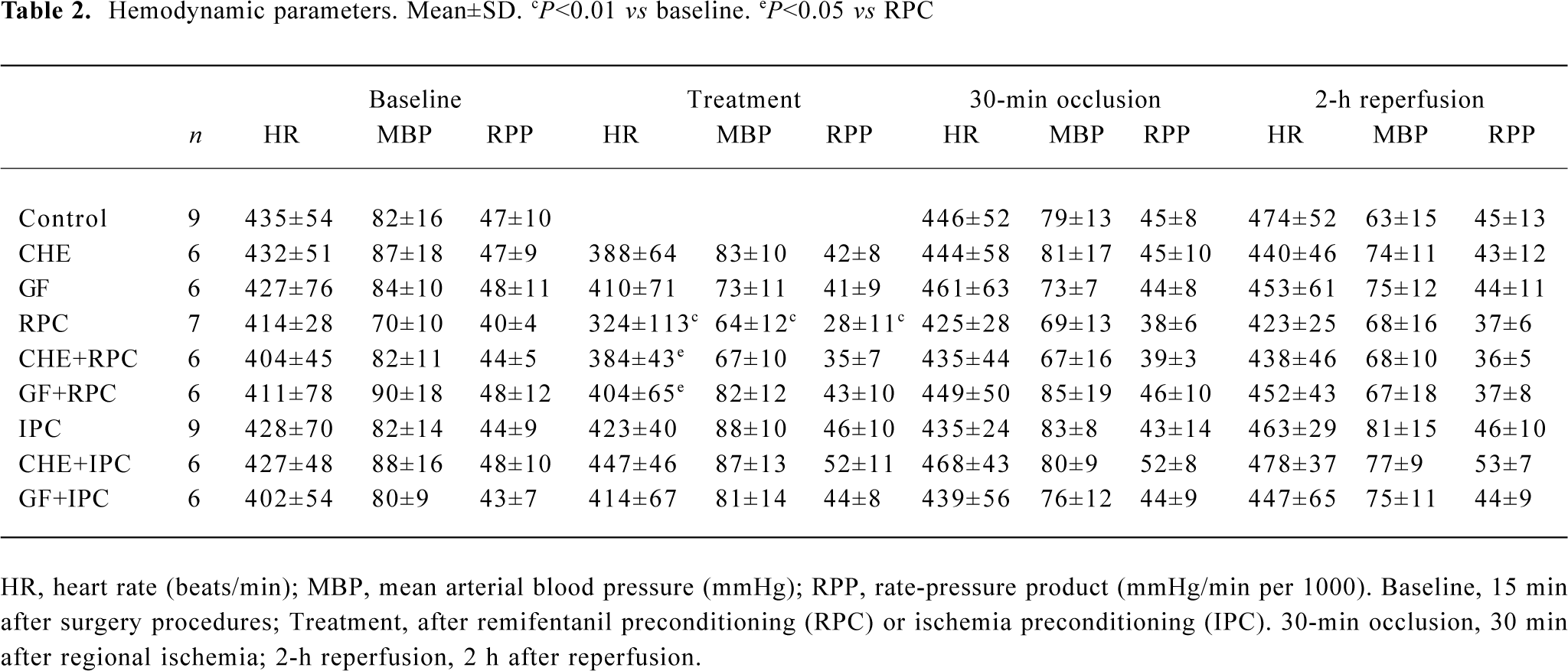
Full table
Discussion
The present results demonstrated that remifentanil conferred cardioprotection against injury induced by ischemic reperfusion, which was completely abolished by CHE and GF, both PKC inhibitors, and suggests that RPC, like IPC, protects myocardium by a mechanism that involves PKC activation.
Miki et al[10] found that the cardioprotective effect of morphine could be blocked by the PKC inhibitor CHE and OR participate in the triggering effect of IPC through activation of PKC. Also, Kato et al[4] suggested that fentanyl limited infarction size through meditation by PKC activation. Our data showed that CHE or GF abolished the protective effect of RPC, suggesting that like morphine and fentanyl, the protective effect of RPC on postischemia myocardial injury is mediated by a PKC activated pathway. In agreement with previous studies, we also found blockade of PKC abolished the effect of IPC.
The cellular mechanisms by which RPC exerts its postischemic protective action are unknown. Ligand-binding data show that remifentanil has a high degree of μ-opioid receptor selectivity (EC50=2.6 nmol/L) with a lower affinity for δ (EC50=66 nmol/L) and κ (EC50 = 6.1 μmol/L) opioid receptors. Previously, we found remifentanil reduced IS dose-dependently in open chest anesthetized rats[5]. The protective effect of RPC was abolished by all three OR antagonists CTOP, naltrindole, and nor-binaltorphinmine, indicating that the effect of remifentanil is mediated via µ-, δ-, and κ-OR[5]. The OR are known to couple to pertussis toxin sensitive G proteins such as Gi or Go[15,16]. If opioid receptors act by activation of PKC, then they must couple to a phospholipse. In the heart, δ- and κ-OR stimulation could increase the level of inositol 1,4,5-triphosphate, suggesting phospholipase C or D-mediated turnover of phosphatidylinositol[17]. Opioids are linked to PKC and are therefore putative mediators in RPC or IPC.
Furthermore, intracellular signaling pathways, which mediate subtype of the OR-induced cardioprotection have been studied previously. Fryer et al[12] found that TAN-67, a selective δ1-OR agonist, had an IS reduction effect that was abolished by CHE and GF, two PKC inhibitors that act on different binging sites on PKC to produce an inhibitory effect. In contrast, our lab showed that the cardioprotection of activation of κ-OR and IPC was significantly attenuated by blockade of PKC with PKC inhibitor CHE in the isolated rat heart[14,18,19]and myocytes[19]. These results provided evidence that the effect of RPC on postischemic hearts was partially mediated via a PKC activated pathway.
The role of PKC in IPC and opioid-induced PC is not fully understood[20,21]. Generally, opioids activate δ-and κ-OR, which lead to PKC activation. Activated PKC acts as an amplifier of the preconditioning stimulus and stabilizes, by phosphorylation, the open state of the mitochondrial KATP channel and the sarcolemmal KATP channel. PKC-δ translation seems to be responsible for activating mitochondrial KATP channels and PKC-ε translocation for the establishment of late preconditioning by phosphorylating nuclear targets. The opening of KATP channels ultimately elicits cytoprotec-tion by decreasing cytosolic and mitochondrial Ca2+ overload[22].
Although our data also show that pretreatment with PKC inhibitor CHE or GF could prevent HR from decreasing led by RPC, the difference was significant among all treatment groups and control. It suggests that PKC is also involved in the bradycardia response of remifentanil.
We conclude that RPC limits infarction in open chest rat hearts via PKC activation mechanism, which mimics the effect of IPC.
Acknowledgments
The authors thank CP MOK for technical assistance.
References
- Schultz JE, Hsu AK, Gross GJ. Morphine mimics the cardioprotec-tive effect of ischemic preconditioning via a glibenclamide-sensitive mechanism in the rat heart. Circ Res 1996;78:1100-4.
- Schultz JJ, Hsu AK, Gross GJ. Ischemic preconditioning and morphine-induced cardioprotection involve the delta (delta)-opioid receptor in the intact rat heart. J Mol Cell Cardiol 1997;29:2187-95.
- Kato R, Ross S, Foex P. Fentanyl protects the heart against ischaemic injury via opioid receptors, adenosine A1 receptors and KATP channel linked mechanisms in rats. Br J Anaesth 2000;84:204-14.
- Kato R, Foex P. Fentanyl reduces infarction but not stunning via delta-opioid receptors and protein kinase C in rats. Br J Anaesth 2000;84:608-14.
- Zhang Y, Irwin M, Wong TM. Remifentanil preconditioning protects against ischemic injury in the intact rat heart. Anesthesiology 2004;101:918-23.
- James MK, Feldman PL, Schuster SV, Bilotta JM, Brackeen MF, Leighton HJ. Opioid receptor activity of GI 87084B, a novel ultra-short acting analgesic, in isolated tissues. J Pharmacol Exp Ther 1991;259:712-8.
- Egan TD, Minto CF, Hermann DJ, Barr J, Muir KT, Shafer SL. Remifentanil versus alfentanil: comparative pharmacokinetics and pharmacodynamics in healthy adult male volunteers. Anesthesiology 1996;84:821-33.
- Schultz JE, Gross GJ. Opioids and cardioprotection. Pharmacol Ther 2001;89:123-37.
- Gross GJ. Role of opioids in acute and delayed preconditioning. J Mol Cell Cardiol 2003;35:709-18.
- Miki T, Cohen MV, Downey JM. Opioid receptor contributes to ischemic preconditioning through protein kinase C activation in rabbits. Mol Cell Biochem 1998;186:3-12.
- Fryer RM, Schultz JE, Hsu AK, Gross GJ. Pretreatment with tyrosine kinase inhibitors partially attenuates ischemic preconditioning in rat hearts. Am J Physiol 1998;275:H2009-15.
- Fryer RM, Wang Y, Hsu AK, Gross GJ. Essential activation of PKC-delta in opioid-initiated cardioprotection. Am J Physiol Heart Circ Physiol 2001;280:H1346-53.
- Schultz JE, Hsu AK, Gross GJ. Ischemic preconditioning in the intact rat heart is mediated by delta1- but not mu- or kappa-opioid receptors. Circulation 1998;97:1282-9.
- Wang GY, Wu S, Pei JM, Yu XC, Wong TM. Kappa- but not delta-opioid receptors mediate effects of ischemic preconditioning on both infarct and arrhythmia in rats. Am J Physiol Heart Circ Physiol 2001;280:H384-91.
- Aghajanian GK, Wang YY. Pertussis toxin blocks the outward currents evoked by opiate and alpha 2-agonists in locus coeruleus neurons. Brain Res 1986;371:390-4.
- Burns DL, Hewlett EL, Moss J, Vaughan M. Pertussis toxin inhibits enkephalin stimulation of GTPase of NG108-15 cells. J Biol Chem 1983;258:1435-8.
- Ventura C, Spurgeon H, Lakatta EG, Guarnieri C, Capogrossi MC. Kappa and delta opioid receptor stimulation affects cardiac myocyte function and Ca2+ release from an intracellular pool in myo-cytes and neurons. Circ Res 1992;70:66-81.
- Cao CM, Xia Q, Tu J, Chen M, Wu S, Wong TM. Cardioprotection of interleukin-2 is mediated via {kappa}-opioid receptors. J Pharmacol Exp Ther 2004;309:560-7.
- Wu S, Li HY, Wong TM. Cardioprotection of preconditioning by metabolic inhibition in the rat ventricular myocyte. Involvement of kappa-opioid receptor. Circ Res 1999;84:1388-95.
- Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res 2004;61:427-36.
- Simkhovich BZ, Przyklenk K, Kloner RA. Role of protein kinase C as a cellular mediator of ischemic preconditioning: a critical review. Cardiovasc Res 1998;40:9-22.
- Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth 2003;91:551-65.