
Effect of hydrogen peroxide on persistent sodium current in guinea pig ventricular myocytes1
Introduction
The persistent sodium current (INa.P) in ventricular myocytes results from inactivate-resistant sodium channels continuing to open for long periods during prolonged depolarization. It plays an important role in maintaining the plateau of the action potential (AP), determining AP duration and transmural dispersion of repolarization, and development of cardiac arrhythmias[1]. Some studies suggest that hypoxia can increase cardiomyocyte INa.P and the potentiated INa.P can induce intracellular calcium overload and ischemic arrhythmias[2,3]. Recently, we studied the mechanisms of INa.P generation and augmentation in normoxic and hypoxic conditions. The results suggest that excess nitric oxide (NO) produced during hypoxia can increase ventricular myocyte INa.P by oxidizing cell membrane sodium channel protein, and the reducing agent dithiothreitiol (DTT) can block the potentiated INa.P completely. This implies that oxidants can increase cardiac INa.P, and that reducing and the antioxidation agents can reduce INa.P[4]. A burst of hydrogen peroxide (H2O2) is generated in the myocardium during ischemia. Its effects on cell damage and membrane lipid peroxidation have been reported widely. However, reports on the effect of H2O2 on INa.P in ventricular myocytes are scarce and contradictory[5,6]. In the present study, we examined the effects of H2O2 on INa.P in guinea pig ventricular myocytes and explored the possible mechanisms underlying them.
Materials and methods
Isolation of guinea pig ventricular myocytes Adult guinea pigs (250 g–300 g, of either sex, Grade II, Certificate N
Electrical recordings Myocytes were transferred to a chamber mounted on the mechanical stage of an inverted microscope (XDS-1, Chongqing, China), and perfused with normal Tyrode’s solution containing: 135 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, 0.33 mmol/L NaH2PO4, 10 mmol/L HEPES, 10 mmol/L glucose, pH 7.4. Patch electrodes were pulled with a 2-stage puller (PP-830, Narishige Group, Tokyo, Japan). For whole-cell recordings, their resistances were in the range of 1.5 MΩ–3 MΩ when filled with a pipetted solution containing: 120 mmol/L CsCl, 1.0 mmol/L CaCl2, 5 mmol/L MgCl2, 5 mmol/L Na2ATP, 10 mmol/L TEACl, 11 mmol/L egtazic acid, and 10 mmol/L HEPES, pH 7.3. The external solution was Tyrode’s solution with CdCl2 (200 µmol/L). For single-channel recordings, the shanks of pipettes with resistance of 6 MΩ–10 MΩ were coated with Sylgard and the tips were heat polished. The pipettes were filled with a solution composed of 180 mmol/L NaCl, 1.3 mmol/L KCl, 1.5mmol/L CaCl2, 0.5 mmol/L MgCl2, 5 mmol/L Na2ATP, 3.0 mmol/L CoCl2, 10 mmol/L TEACl, 10 mmol/L 4-AP, 10 mmol/L CsCl, 5 mmol/L HEPES, and 5 mmol/L glucose, pH 7.4. For single-channel cell-attached recordings, the myocytes were perfused with normal Tyrode’s solution. For single-channel inside-out recordings, the perfusate was composed of 120 mmol/L KCl, 0.1 mmol/L CaCl2, 2.0 mmol/L MgCl2, 0.1mmol/L egtazic acid, and 10 mmol/L HEPES, pH 7.4. These perfusates were bubbled with 100% O2. All experiments were carried out at room temperature (21±2 oC). Currents were obtained with a patch-clamp amplifier (EPC-9, Heka Electronic, Lambrecht, Pfalz, Germany), filtered at 2 kHz, digitized at 10 kHz, and stored on a computer hard disk for further analysis.
Drugs and reagents Collagenase type I and CsCl were obtained from Gibco (GIBCO TM, Invitrogen Co., Paisley, UK). Protease E, 4-AP, TEACl, Na2ATP, and egtazic acid were purchased from Sigma Chemical Co (Saint Louis, Missouri, USA). Bovine serum albumin, HEPES, and taurine were obtained from Roche(Basel, Switzerland). Tetrodotoxin (TTX) was purchased from Hebei Fisheries Research Institute (Qinhuangdao, China). H2O2 was a production of Wuhan Zhongnan Chemical Reagent Co (Wuhan, China). Reduced GSH was a product of Shanghai Bio Life Science and Technology (Shanghai, China).
Data analysis Whole-cell recordings were analyzed using PulseFit (V8.65, HEKA). Current density was calculated by dividing the current amplitude by the cell capacitance. Single-channel recordings were analyzed using TAC+TACFit (X4.0.9, Bruxton, Seattle, Washington, USA). Capacitance transients and leakage currents were nullified by off-line subtracting fits of average blunt traces. The channel activity after a 50 ms depolarization pulse was calculated as persistent sodium channel. Open probability was calculated from the total open times of 50 sweeps divided by the total sweep duration. Histograms of channel open time distribution were fitted to single exponentials using TACFit. All data were expressed as mean±SD. Student’s t-test was used for simple comparisons and ANOVA was used when appropriate. P<0.05 was considered to be statistically significant.
Results
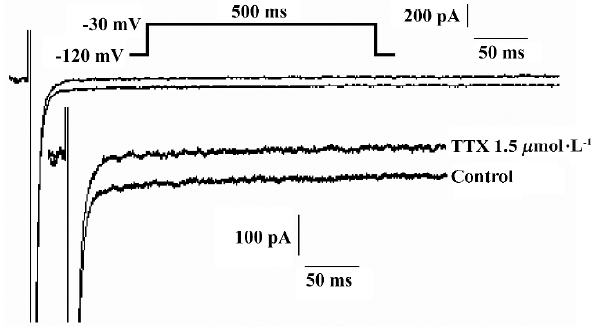
H2O2 increases the amplitude of INa.P in a concentration-dependent manner The experiments were carried out in the whole-cell configuration of the patch-clamp techniques. Currents were evoked by a 500 ms pulse to -30 mV from a holding potential of -120 mV. TTX (1.5 µmol/L) blocked completely the smaller persistent inward current, but had less effect on the transient sodium current (INa.T), (n=7 cells from 4 guinea pigs; Figure 1). The recorded current was proved to be INa.P.
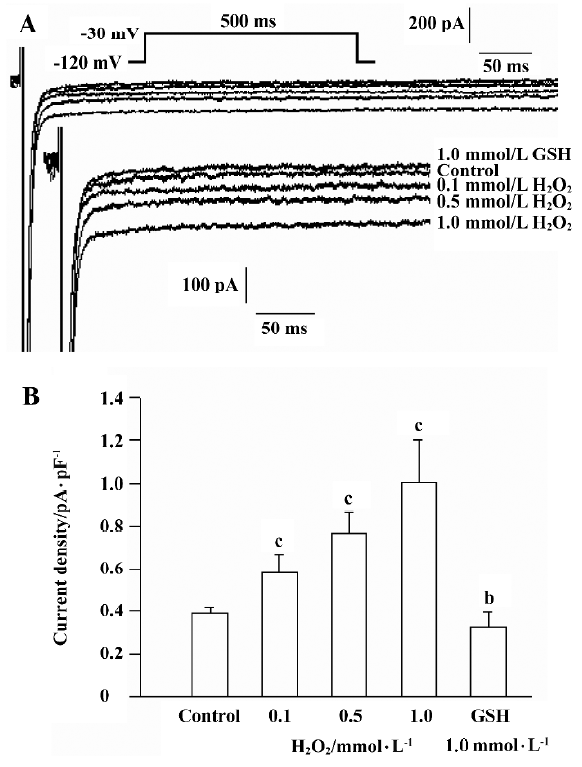
Using the pulse protocol described above, 4 cells were perfused with H2O2 (0.5 mmol/L) after the INa.P had stabilized. INa.P began to increase at approximately 5 min and reached a maximum at 10−12 min after perfusion with H2O2. The current is able to last for another 30 min at this level. In another 8 cells, H2O2 was added into solution in a cumulative manner after the control INa.P values were recorded. Cells were perfused with H2O2 (0.1 mmol/L, 0.5 mmol/L and 1.0 mmol/L) and GSH (1.0 mmol/L) at 10 min intervals, and the currents were recorded in the same cell. H2O2 increased the amplitude of INa.P in a concentration-dependent manner, while GSH reversed the increased INa.P (Figure 2). The amplitude of INa.P was recorded at 200 ms of the pulse to eliminate the effect of INa.T. H2O2 (0.1 mmol/L, 0.5 mmol/L, and 1.0 mmol/L) increased the mean current density of INa.P from the control value 0.385±0.032 pA/pF to 0.578±0.080 pA/pF, 0.763±0.094 pA/pF, and 1.007±0.179 pA/pF, respectively (n=8, P<0.01 vs control). The mean current density was decreased to 0.329±0.063 pA/pF after the application of 1.0 mmol/L GSH (n=8, P<0.05 vs control).
Effect of glutathione on persistent sodium channel activity induced by H2O2 The experiments were carried out in cell-attached and inside-out patches. Currents were activated by a 700 ms voltage pulse to -50 mV from a holding potential of -120 mV.
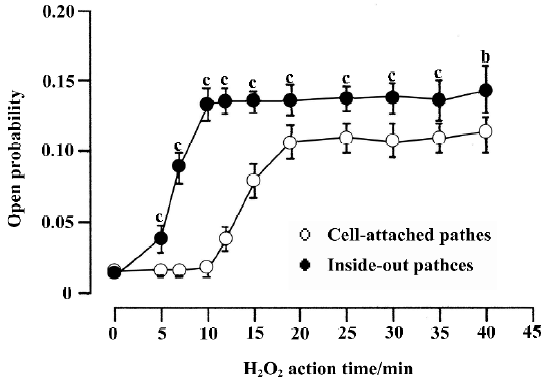
In cell-attached recording (the resting membrane potential was -74.6±7.7 mV, n=12, which was measured in current-clamp mode under identical conditions), bath solution with 1 mmol/L H2O2 was used to perfuse the cell. In 17 cell-attached patches, persistent sodium channel activity increased remarkably at 11.5±3.9 min. The current type was changed from background currents to burst currents (flaring up rapidly but subsiding over a very long period). At 18.7 ± 4.7 min the channel activity reached a maximum. In 6 of the 17 patches, the channel activity could maintain for another 30 min at this level (Figure 3).
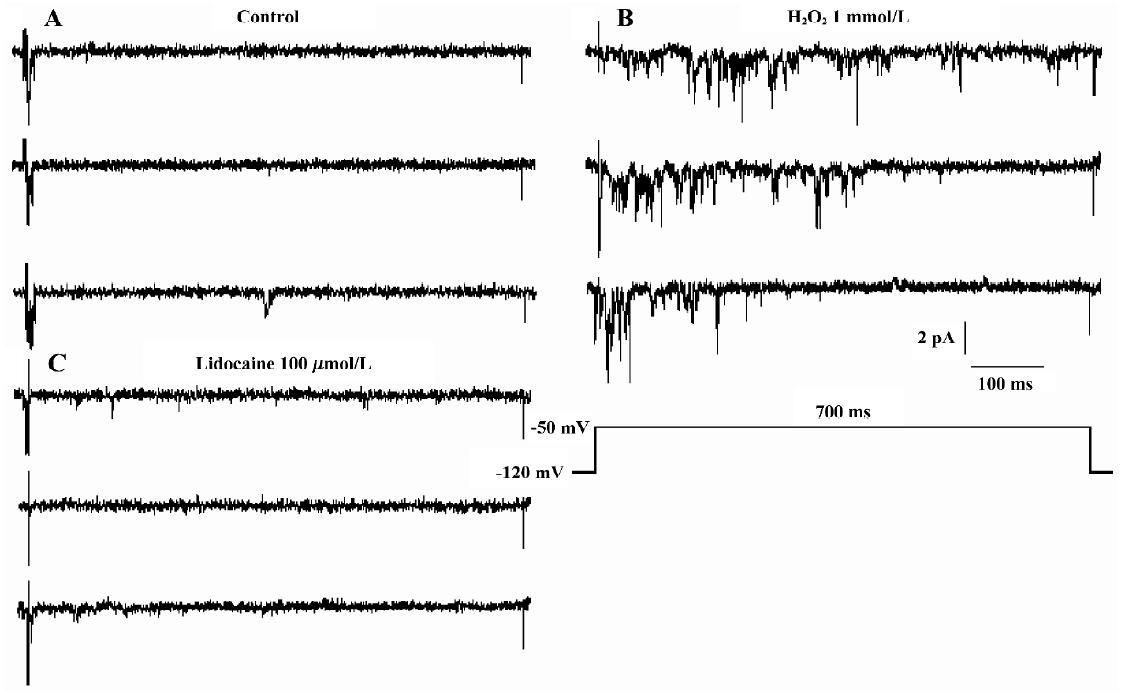
In 5 of the 17 patches, lidocaine (100 µmol/L) was added to the bath solution and the increased INa.P was blocked completely at approximately 5 min (Figure 4).
We applied 1 mmol/L GSH in 6 other patches. The presentation traces are shown in Figure 5. Before exposure to H2O2, channel activity was very low or often absent (Figure 5A). After 10-min treatment with H2O2, persistent sodium channel activity increased markedly (Figure 5B) and reached a maximum at 20 min (Figure 5C). GSH (1 mmol/L) reversed the increase in sodium channel activity caused by H2O2 (Figure 5D). H2O2 (1 mmol/L) increased the mean open probability and mean open time from control values of 0.015±0.004 and 0.744±0.190 ms to 0.106±0.011 and 1.966±0.539 ms, respectively (n=6, both P<0.01 vs control). They were then decreased to 0.039±0.024 and 1.137±0.153 ms, respectively, after the application of 1 mmol/L GSH (n=6, both P<0.01 vs H2O2). Figure 6 gives an example of corresponding all-time histograms and mean open-time histograms from another cell-attached patch.
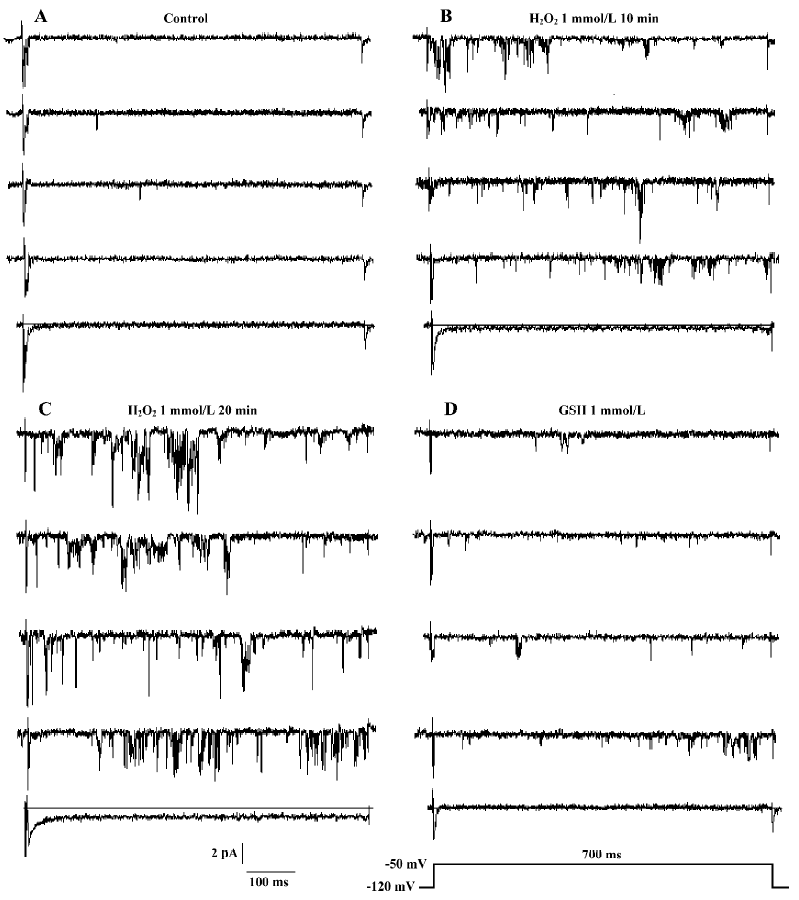
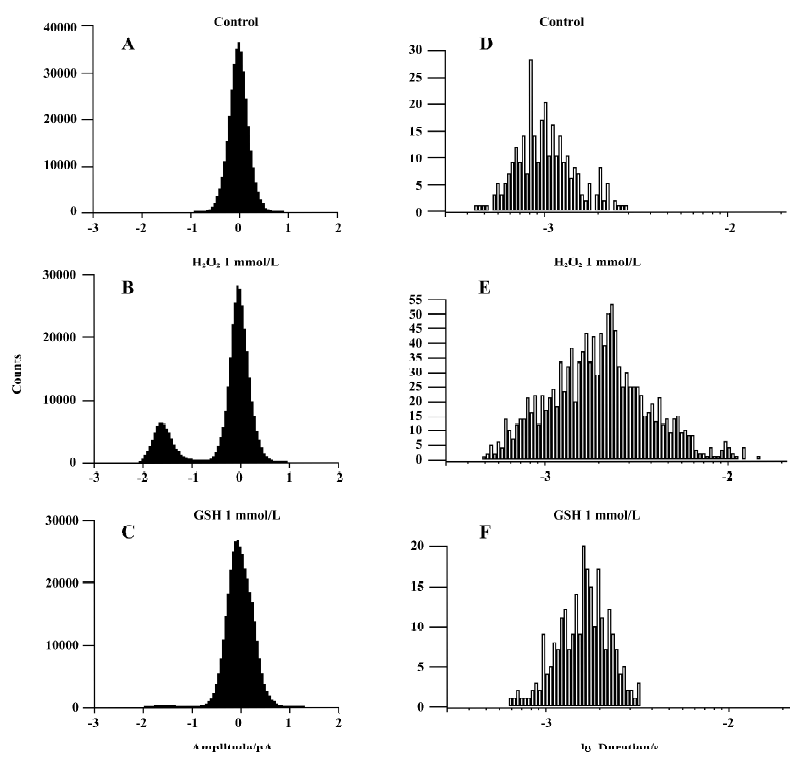
The results for inside-out recordings were similar to those for cell-attached recordings. In the 10 inside-out patches, persistent sodium channel activity increased markedly at 4.8±1.0 min and reached maximum at 9.6±1.6 min after the application of 1 mmol/L H2O2. Compared with the cell-attached recordings, the times when the sodium channel activity in inside-out recordings began to increase (4.8±1.0 min vs 11.5±3.9 min, P<0.01) and reached a maximum (9.6±1.6 min vs 18.7±4.7 min, P<0.01) were shorter. In 5 of the 10 patches, the channel activity could be maintained for another 30 min at this level (Figure 3). In the other 5 patches, 1 mmol/L GSH was applied. H2O2 (1 mmol/L) increased the mean open probability and mean open time from the control value 0.012±0.003 and 0.537±0.015 ms,respec-tively, to 0.136±0.010 and 0.966±0.130 ms, respectively (n=5, both P<0.01 vs control). They were decreased to 0.027±0.006 and 0.672±0.042 ms, respectively, after the application of 1 mmol/L GSH (n=5, both P<0.01 vs H2O2).
Discussion
A large number of studies have reported that H2O2 in cardiomyocytes is increased during ischemia. The excessive amount of H2O2 leads to intracellular Ca2+ overload and cell damage. Recent reports show that hypoxia can increase INa.P in ventricular myocytes, induces intracellular sodium overload, which promotes Ca2+ overload via reverse Na+-Ca2+ exchange, and prolongs the duration of action potential (AP) after-depolarization[2,3]. Thus, the present study on the effect of H2O2 on INa.P is very important to further understand the mechanisms of cardiomyocytes injury induced by H2O2, and the nature of INa.P.
In whole-cell patch-clamp recordings, 1.5 µmol/L TTX blocked completely the smaller inactivation-resistant inward current (Figure 1). Similarly, in cell-attached recordings, 100 μmol/L lidocaine blocked the potentiated inward currents induced by H2O2 (Figure 4). These results affirm that the recorded currents are INaP. Barrington et al reported that there were no effects from a 30-min exposure to 1 mmol/L H2O2 on the slowly inactivating sodium currents of feline ventricular myocytes, so hydroperoxide could not induce an intracellular sodium overload[5]. However, our study shows that H2O2 increases the amplitude of whole-cell INa.P of guinea pig ventricular myocytes in a concentration-dependent manner (Figure 2) and the activity of persistent sodium channel in both single-channel recordings (Figures 3–6). These results confirm that H2O2 can increase INa.P of guinea pig ventricular myocytes, which is different from the report of Barrington et al. The disparity may be attributable to species differences. The result that H2O2 increases INa.P in a concentration-dependent manner is similar to our previous report that hypoxia increases the INa.P of guinea pig ventricular myocytes in a time-dependent manner[4], further suggesting that the increase in INa.P is closely associated with reactive oxygen species. Ward and Giles observed that H2O2 produced a marked prolongation of the AP by slowing inactivation of the TTX-sensitive sodium current, which was verified by the single-channel cell-attached recording results that late opening events were enhanced when 200 µmol/L H2O2 was included in the recording pipette[6]. Bisindolyl-amaleimide, the protein kinase C (PKC) blocker, significantly delayed and attenuated the development of AP prolongation, which indicated involvement of an intracellular second messenger-PKC pathway[6]. In our study, H2O2 increased the persistent sodium channel activity in both the inside-out and cell-attached patches, which suggests that H2O2 may take effect by directly oxidizing the cell membrane in addition to its involvement as an intracellular second messenger. These results are consistent with the findings of Hammarström and Gage who reported that hypoxia, NO, and sodium cyanide could increase persistent sodium channel activity in rat hippocampal neurons in inside-out recordings, which could then be reversed by DTT[7,8].
H2O2 increased the persistent sodium channel activity in both cell-attached and inside-out patches (Figure 3). However, the time when the persistent sodium channel activity began to increase and reached a maximum was significantly shorter in inside-out patches compared with cell-attached patches. One possible explanation is that the inside-out patches, which were directly exposured to H2O2, lost the protection of intracellular antioxidant enzymes and antioxidants such as superoxide dismutase, catalase, glutathione peroxidase, ascorbic acid, α-tocopherol and glutathione.
Glutathione inhibited the H2O2-elicited persistent sod-ium channel activity (Figures 2,5,6), which suggested that H2O2 could take effect by oxidation. It is similar to our previous report, which shows that excessive NO produced during hypoxia can increase the INa.P of ventricular myocytes by oxidizing cell membrane sodium channel proteins and generating INaP under normoxic conditions, probably in association with the oxidation state of channel proteins[4]. H2O2 is a cysteine-specific oxidant[9] and GSH is an important antioxidant that can protect the protein’s thiol group from oxidation[10]. The present study shows that GSH can reverse the persistent sodium channel activity caused by H2O2. Therefore, we think that persistent sodium channel activity may be associated with oxidation state, and oxidants can increase activity by oxidizing the channel protein. H2O2 as an oxidant may increase persistent sodium channel activity by oxidizing the thiol group of proteins in cell membranes.
References
- Antzelevitch C. Electrical heterogeneity, cardiac arrhythmias, and the sodium channel. Circ Res 2000;87:910-4.
- Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol 1996;497:337-47.
- Hammarström AKM, Gage PW. Hypoxia and persistent sodium current. Eur Biophys J 2002;31:323-30.
- Ma JH, Wang XP, Zhang PH. Mechanisms on nitric oxide increasing persistent sodium current of ventricular myocytes in guinea pig during normoxia and hypoxia. Acta Physiol Sin 2004;56:603-8.
- Barrington PL, Martin RL, Zhang K. Slowly inactivating sodium currents are reduced by exposure to oxidative stress. J Mol Cell Cardiol 1997;29:3251-65.
- Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol 1997;500:631-42.
- Hammarström AKM, Gage PW. Nitric oxide increases persistent sodium current in rat hippocampal neurons. J Physiol 1999;520:451-61.
- Hammarström AKM, Gage PW. Oxygen-sensing persistent sodium channels in rat hippocampus. J Physiol 2000;529:107-18.
- Prasad M, Goyal RK. Differential modulation of voltage-dependent K+ currents in colonic smooth muscle by oxidants. Am J Physiol 2004;286:C671-82.
- Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta 2003;333:19-39.