
Isoflurane preconditioning protects against ischemia-reperfusion injury partly by attenuating cytochrome c release from subsarcolemmal mitochondria in isolated rat hearts1
IntroductionOther Section
Volatile anesthetics can protect against reperfusion injury after myocardial ischemia in vitro and in vivo[1–4]. And isoflurane is one of the drugs most commonly used to maintain the state of general anesthesia. Isoflurane pre-conditioning, a temporary exposure to isoflurane followed by its complete washout, can protect against cardiac ischemia- reperfusion injury. It has been reported that isoflurane preconditioning can mimic ischemia preconditioning to protect against reperfusion injury after myocardial ischemia in vitro and in vivo [3,4]. However, to date, the mechanism of isoflurane preconditioning remains unclear. It has been reported that ischemic preconditioning can induce mitochondrial tolerance by using the loss of cytochrome c as an indication of mitochondrial dysfunction[5]. It is not known whether the cardioprotective effect of isoflurane preconditioning can also be achieved by inducing mitochondrial tolerance.
Mitochondria sustains progressive damage during ischemia. Cardiac mitochondria exist in two functionally distinct populations. Subsarcolemmal mitochondria (SSM) are located beneath the plasma membrane, whereas interfibrillar mitochondria (IFM) are located between the myofibrils. These two mitochondria are affected differently in myocardiopathy and ischemia. The progression of ischemic damage is more rapid in SSM[6,7]
In the present study, we examined if isoflurane preconditioning protected against a detrimental ischemic insult by attenuating cytochrome c release from the inner membrane of subsarcolemmal mitochondria
Materials and methodsOther Section
Isolated heart preparation and measurements Experiments were performed on adult male Sprague-Dawley rats weighing 230–300 g. The rats were housed in plastic cages with soft bedding and free access to food and water under a 12-h day/12-h night cycle. Rats were supplied by the Experimental Animal Center of Xuzhou Medical College. All experiments were approved by the Animal Care and Use Committee at the college and were in accordance with the Guidelines for Care and Use of Laboratory Animals. Pentobarbital 40 mg/kg and heparin 500 U/kg were injected intraperitoneally into 44 Sprague-Dawley rats weighing 250–300 g. After thoracotomy, the hearts were isolated and immediately placed in 4 °C Krebs-Henseleit (K-H) solution, then rapidly prepared using the Langendorff method and perfused at 55 mmHg with K-H solution at 37 °C. The perfusate was equilibrated with mixed gas of 95% O2 and 5% CO2 to meet standards (pH, 7.4±0.02; carbon dioxide partial pressure, 25±4 mmHg; oxygen partial pressure, 570±20 mmHg). The composition of K-H perfusate was as follows: NaCl 118 mmol/L, KCl 4.7 mmol/L, KH2PO4 1.2 mmol/L, CaCl2 2.5 mmol/L, MgSO4 1.2 mmol/L, NaHCO3 25 mmol/L, glucose 11.1 mmol/L, Na2-EDTA 0.125 mmol/L.
Left ventricular pressure (LVP) and dp/dt were measured isovolumetrically with a saline-filled latex balloon inserted into the left ventricle through a cut in the left atrium. At the beginning of the experiment, the balloon volume was adjusted to achieve a diastolic LVP of 0 mmHg, so that any subsequent increase in diastolic LVP indicated an increase in left ventricular wall stiffness or diastolic contracture. Baseline hemodynamics were recorded 20 min after stabilization. Hearts were randomly assigned to a non-treated group (CON group, n=12) or three isoflurane preconditioning groups (0.5% ISC group, 1.0% ISC group and 2.0% ISC group; n=12) and were perfused with K-H solution saturated with 0.0%, 0.5%, 1.0%, and 2.0% isoflurane, respectively, for 15 min and followed by a 15-min washout with normal K-H solution before 30 min ischemia and 60 min reperfusion. Isoflurane was bubbled into the perfusate with an isoflurane vaporizer (Ohmeda, West Yorkshire, England) placed in the oxygen-carbon dioxide gas mixture line. Vapour concentrations were measured continuously by an anesthetic gas detector (Capnomac; Ultima, Datex-Engstrom, Helsinki, Finland). LVP, dp/dt, and HR were monitored continuously during the whole experiment using Maclab 4.0 software (AD Instru-ments, Australia).
Measurement of infarct size The 2,3,5-triphenyltetrazo-lium chloride (TTC) staining technique was used to determine infarct size after 60-min reperfusion. Fresh TTC was prepared in 0.1 mol/L phosphate buffer adjusted to pH 7.4. TTC stained the non-infarcted myocardium a bright red color, caused by reduction of TTC by dehydrogenases present in viable tissues. Hearts stored at -70 °C after each experiment were taken up and sliced into 4–6 transverse sections (3-mm thickness). The sections were immersed in 1% TTC solution and incubated for 30 min at 37 °C. All slices were digitally imaged by a photoscanner[8], and the infarcted areas of each slice were measured in a blinded fashion by planimetry using Imagemaster VDS (Pharmacia Bioter, USA).
Isolation of subsarcolemmal rat heart mitochondria and cytosolic fractions Subsarcolemmal mitochondria were isolated from the ventricles as follows: the ventricles were rapidly dissected and rinsed in ice-cold homogenization buffer (CP1: KCl 100 mmol/L, MOPS 50 mmol/L, MgSO4 5.0 mmol/L, edetic acid 1.0 mmol/L, ATP 1.0 mmol/L, at pH 7.4). The ventricles were blotted dry, weighed, minced, and washed with CP1. The mince was homogenized at 20 mL/g tissue in cold CP2 (CP1+BSA 2.0 g/L). The homogenate was then centrifuged at 584×g for 10 min. The supernatant was filtered through double layer gauze and centrifuged at 3015×g for 10 min. The mitochondrial pellet was resuspended in 5 mL CP2 per gram of heart and centrifuged at 3015×g for 10 min, and finally subsarcolemmal mitochondria were resuspended in 2.5 mL KME (100 mmol/L KCl, 50 mmol/L MOPS, 5 mmol/L egtazic acid) per gram of heart and centrifuged at3 015×g for 10 min. Subsarcolemmal mitochondria was resuspended into KME at a final protein concentration of approximately 1.5 g/L. The first 3015×g supernatant represented the cytosolic fraction (protein concentration of approximately 2.0 g/L). All manipulations were carried out at 4 °C. Protein concentration was determined using Bradford assay, with BSA as standard.
Detection of cytochrome c by Western blotting Mitochondria prepared from isolated rat hearts were added 0.25% volume of 2×sample buffer (250 mmol/L Tris-HCl, 8.0% SDS, 700 mmol/L sucrose, 300 mmol/L DTT, 0.01% bromophenol blue, at pH 6.8). Samples were loaded onto 15% Tris-buffered polyacrylamide gels and component proteins resolved by SDS-PAGE. Proteins were electroblotted onto a nitrocellulose membrane and cytochrome c was detected using monoclonal anti-cytochrome c (Santa Cruz Biotechnology, Inc, USA) as primary antibody and anti-mouse IgG conjugated to alkaline phosphatase as secondary antibody. Primary antibody binding was visualized with an alkaline phosphatase based chemiluminescence system.
Electron microscopy Mitochondria isolated from the myocardium in several kinds of buffer by density centrifugation was resuspended and fixed with 2% glutaraldehyde in 0.1 mol/L PBS buffer. Mitochondria were post-fixed using 1% OsO4. En bloc staining with uranyl acetate was followed by dehydration and embedding. Embedded samples were sectioned and affixed to grids according to standard protocols. Mitochondrial ultrastructure was then evaluated by transmission electron microscopy.
Statistical analysis All data were expressed as mean± SD. Statistical analysis of data within and between groups was performed with analysis of variance (ANOVA) for repeated measures followed by Turkey multiple-comparison post-hoc test. P<0.05 was considered statistically significant.
ResultsOther Section
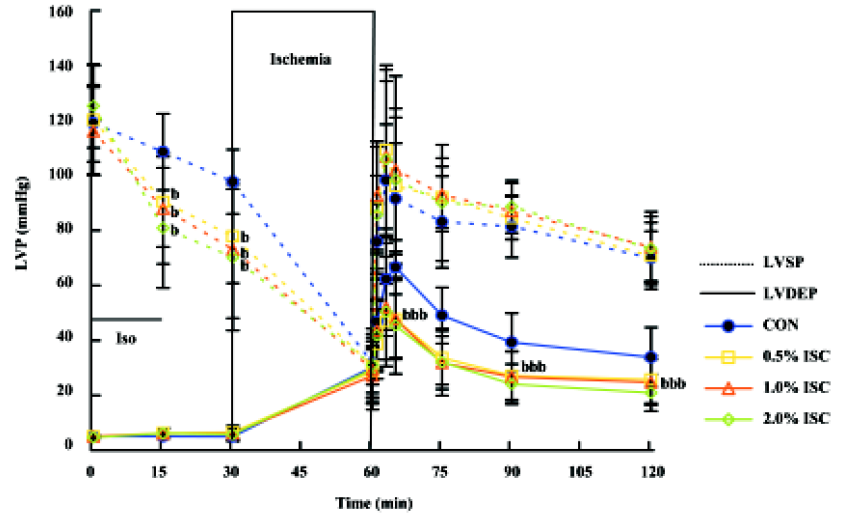
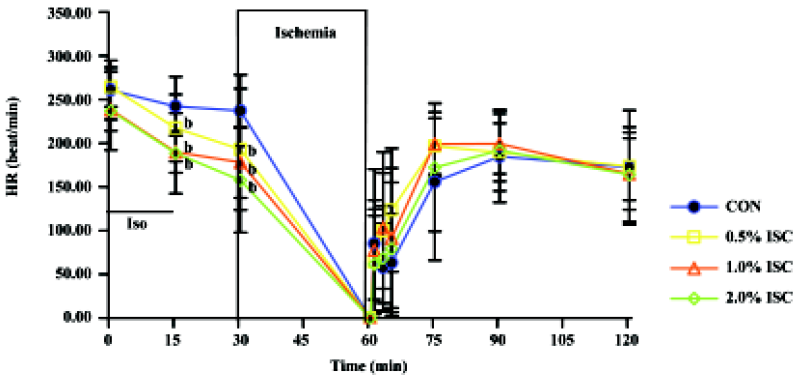
Changes in hemodynamics Figures 1,2,3 show changes in LVP, dp/dtmin, dp/dtmax, and HR of hearts in each of the four groups during the time course of the experiment. There was no difference in baseline hemodynamics among experimental groups. A concentration-dependent depression of left ventricular systolic pressure (LVSP), dp/dtmax, and heart rate was observed after 15 min of isoflurane treatment. After 60 min of reperfusion, hemodynamic function decreased in each group compared with baseline values. Left ventricular end diastolic pressure (LVEDP) was lower in the three isoflurane-preconditioning groups than in the CON group at 30 and 60 min reperfusion, but there was no significant difference in LVSP, dp/dtmin, dp/dtmax, and HR among the four groups.

Infarct size Figure 4 shows that the infarct size of the CON group was 56%±12%, isoflurane-preconditioning (0.5%,1.0%, and 2.0%) significantly reduced infarct size to 41%± 12%, 32%±7%, and 33%±11%, respectively (P<0.05). But there was no significant difference among the three isoflurane-preconditioning groups.

Morphology of isolated mitochondria Electron microscopy analysis (×10 000) reveals basically formed membranes and clearly discernable cristae in the mitochondria isolated from hearts. The outer membranes of mitochondria isolated from CON groups were partly disrupted, and mitochondria were severely swollen, with fragmentation of the cristae. Isoflurane preconditioning attenuated morphological changes of mitochondria after ischemia and reperfusion. Mitochondria in the 2% isoflurane-preconditioned group appeared to be morphologically better than those of the other preconditioning groups (Figure 5).

Western blot analysis of the release of mitochondrial cytochrome c Isoflurane-preconditioning reduces ischemia reperfusion-induced cytochrome c release from the mitochondria in a concentration-related manner. The results showed the amounts of cytosolic cytochrome c significantly decreased (P<0.05 vs CON group) in an isoflurane concentration-related manner, while the amounts of mitochondrial cytochrome c markedly increased (P<0.05 vs CON group). The results indicate that mitochondrial dysfunction occurred after reperfusion and was attenuated by isoflurane-preconditioning (Figure 6).

DiscussionOther Section
In the present study, by Western blot examining of cytochrome c in mitochondria and cytosol, we observed that isoflurane increased the content of cytochrome c in mitochondria while reduced it in cytosol in a concentration-dependent manner. Isoflurane at concentration of 2% showed the most significant effect on cytochrome c loss in the mitochondria. Furthermore, mitochondria isolated from three isoflurane preconditioning groups were morphologically better than that isolated from CON. Similarly, isoflurane preconditioning significantly reduced the infarct size of hearts at the end of reperfusion. Therefore, we concluded that cytochrome c release from the mitochondria played a key role in the pathogenesis of ischemia/reperfusion injury and myocardioprotective effects of isoflurane preconditioning were associated with the attenuation of cytochrome c loss from the inner membrane of subsarcolemmal mitochondria.
Cytochrome c is a 12-kDa protein which functions in the mitochondrial electron transport chain. At physiological ionic strength, cytochrome c diffuses in the aqueous phase between the inner and outer membranes (outer compartment)and between complex III (cytochrome bc1) and complex IV (cytochrome aa3). But as a small and water-soluble molecule, cytochrome c is easier to be released from mitochondria than other mitochondrial proteins. Studies have shown that the loss of cytochrome c from mitochondria could occur during ischemia and after reperfusion[9,10]. Ischemia tolerance induced by sublethal ischemia is associated with mitochondrial protection (attenuating cytochrome c release from mitochondria) in vitro and in vivo[5].
Increasing evidence suggests that lethal reperfusion injury possibly consists of two forms of cell death, necrosis and apoptosis. The apoptotic process is initiated shortly after the onset of ischemia, and becomes markedly enhanced during reperfusion. Inhibition of the apoptotic process can then attenuate irreversible injury in connection with reperfusion[11]. One of the main mechanisms of cellular death induced by ischemia/reperfusion appears to be mitochondrial dysfunction. The cytochrome c release from mitochondria is a rapid and apoptosis-specific process within 1 h after the induction of apoptosis[12]. In the process of apoptosis, cytochrome c is released from the mitochondria to cytosol and caspase-3 is activated [13]. At the same time, the loss of cytochrome c could also lead to the formation of free radicals, and disturbances of oxidative phosphorylation. Whether the release of cytochrome c leads to necrotic or apoptotic cellular death depends largely on intracellular ATP levels. Most of this protein release from mitochondria to cytosol results in ATP depletion associated with necrotic cell death. Therefore, loss of cytochrome c is considered to be an indication of mitochondrial dysfunction[14,15]. Our results show a possible involvement of mitochondria in the cellular death pathway that is initiated during ischemia/reperfusion. Compared with the other groups, more cytochrome c of CON group was released from mitochondria to cytosol. Also the infarct size in the CON group was higher than those of the isoflurane preconditioning groups. Isoflurane preconditioning attenuated cytochrome c release along with reduced infarct size. Our study demonstrates for the first time in isolated hearts that isoflurane preconditioning is able to attenuate cytochrome c release from mitochondria in a concentration-dependent manner. Meanwhile, isoflurane preconditioning significantly deceased infarct size and morphologically ameliorated mitochondria injury suffered from ischemia and reperfusion. This result also indicated that the redistribution of cytochrome c was highly correlated with cellular death.
The mechanism by which isoflurane preconditioning induces mitochondrial tolerance via attenuating cytochrome c loss is not clear. The effect of preconditioning may first be on cytosolic factors, which lead to mitochondrial tolerance that persists until mitochondria were isolated for testing. Mitochondrial ATP-sensitive potassium (KATP) channels appear to be involved in the process of anesthetics-preconditioning[15–21]. Recent studies have shown that activation of KATP channels reduces the loss of mitochondrial cytochrome c through the protein kinase C signaling[22]. Anesthetic-induced preconditioning reduced cytosolic Ca2+ loading, one factor of cytochrome c release, in part through mitochondrial KATP channels[15,23]. Furthermore, Bcl-2 family proteins are important endogenous factors in regulating cytochrome c release from mitochondria, in which antiapoptotic proteins (eg, Bcl-2, Bcl-XL, etc) inhibit, but proapoptotic proteins (eg, Bax, Bad, etc) enhance, cytochrome c release[24,25]. Therefore, the expression and redistribution of Bcl-2 family proteins is possibly another important factor of isoflurane preconditioning inducing mitochondria tolerance by attenuating cytochrome c release. There is evidence that ischemia and some pharmacological preconditioning are able to induce transcription and expression of Bcl-2 and Bcl-XL without changing transcription and the expression of Bax[26–28].
In the present study, isoflurane preconditioning reduced cytochrome c loss from mitochondria in a concentration-dependent manner. There was a remarkable reduction in cytochrome c release associated with higher concentrations of isoflurane preconditioning. However, infarct size was no further reduced by 2.0% isoflurane, which indicated that isoflurane preconditioning reduced infarct size with a peak effect at a concentration of approximately 1%. Although the release of cytochrome c from mitochondria is the main pathway of cellular death, it is not the only one. For example, members of the death receptor family, such as the Fas receptor and the tumor necrosis factor receptor also play a role in cellular death. Isoflurane preconditioning significantly attenuated cytochrome c release, but the effects of isoflurane preconditioning on other cell death signaling pathways is unclear. It is possible that other pathways of inducing cellular death contributed to the myocardial infarct in 2.0% ISC group. In the present study, however, it can be inferred that isoflurane preconditioning can protect against myocardial ischemic/reperfusion injury in part through attenuating cytochrome c loss. At the same time, it is not appropriate to tell directly whether the cytochrome c release from mitochondrial is likely to be a final step or trigger factor of myocardial injury from the present study. Further studies should be taken to examine if blocking of isoflurane preconditioning could abolish the attenuated release of cytochrome c.
In conclusion, the present study demonstrated that isoflurane preconditioning was capable of protecting against myocardium ischemia/reperfusion injury in part by attenuating the release of cytochrome c from the mitochondria.
AcknowledgementOther Section
The authors are grateful to Dr Qun CHEN for his helpful instruction on the experiment.
ReferencesOther Section
- Kehl F, Krolikowski JG, Mraovic B, Pagel PS, Warltier DC, Kersten JR. Is isoflurane-induced preconditioning dose related? Anesthesiology 2002;96:675-80.
- Obal D, Preckel B, Scharbatke H, Miillenheim J, Hoterkes F, Thamer V, et al. One MAC of sevoflurane provides protection against reperfusion injury in the rat heart in vivo. Br J Anaesth 2001;87:905-11.
- Martini N, Preckel B, Thämer V, Schlack W. Can isoflurane mimic ischemia preconditioning in isolated rat? Br J Anaesth 2001;86:269-71.
- Belhomme D, Peynet J, Louzy M, Launay JM, Kitakaze M, Menasché P. Evidence for precondition by isoflurane in coronary artery bypass graft surgery. Circulation 1999;100:II–340.
- Zhan RZ, Fujihara H, Baba H, Yamakura T, Shimoji K. Ischemic preconditioning is capable of inducing mitochondrial tolerance in the rat brain. Anesthesiology 2002;97:896-901.
- Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol 1997;273:H1544-54.
- Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 2001;280:H2770-8.
- Riess ML, Novalija E, Camara AKS, Eells JT, Chen Q, Stowe DF. Preconditioning with sevoflurane reduces changes in nicotinamide adenine dinucleotide during ischemia-reperfusion in isolated hearts. Anesthesiology 2003;98:387-95.
- Soeda J, Miyagawa S, Sano K, Masumoto J, Taniguchi SI, Kawasaki S. Cytochrome c release into cytosol with subsequent caspase activation during warm ischemia in rat liver. Am J Physiol Gastrointest Liver Physiol 2001;281:G1115-23.
- Czerski LW, Szweda PA, Szweda LI. Dissociation of cytochrome c from the inner-mitochondrial membrane during cardiac ischemia. J Biol Chem 2003;278:34499-504.
- Ji ES, Yue H, Wu YM, He RR. Effects of phytoestrogen genistein on myocardial ischemia/reperfusion injury and apoptosis in rabbits. Acta Pharmacol Sin 2004;25:306-12.
- Renz A, Berdel WE, Kreuter M, Bleka C, Schulze-Osthoff K, Los M. Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivo. Blood 2001;98:1542-8.
- Hu XM, Zhang Y, Zeng FD. Effects of β-aescin on apoptosis induced by transient focal cerebral ischemia in rats. Acta Pharmacol Sin 2004;25:1267-75.
- Shiraishi J, Tatsumi T, Keira N, Akashi K, Mano A, Yamanaka S, et al. Important role of energy-dependent mitochondrial pathways in cultured rat cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol 2001;281:H1637-47.
- Leist BM, Single B, Castoldi AF, Kühnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 1997;185:1481-6.
- Chen Q, Camara A, An JZ, Novalija E, Riess ML, Stowe DF. Sevoflurane preconditioning before moderate hypothermic ischemia protects against cytosolic [Ca2+] loading and myocardial damage in part via mitochondrial KATP channels. Anesthesiology 2002;97:912-20.
- Rourke BO. Myocardial KATP channels in preconditioning. Circ Res 2000;87:845.
- Shimizu J, Sakamoto A, Ogawa R. Activation of the adenosine triphosphate sensitive mitochondrial potassium channel is involved in the cardioprotective effect of isoflurane. J Nippon Med Sch 2001;68:238-44.
- Tanaka K, Weihrauch D, Ludwig LM, Kersten JR, Pagel PS, Warltier DC. Mitochondrial adenosine triphosphate-regulated potassium channel opening acts as a trigger for isoflurane-induced preconditioning by generating reactive oxygen species. Anesthesiology 2003;98:935-43.
- Lim KH, Javadov SA, Das M, Clarke SJ, Suleiman MS, Halestrap AP. The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J Physiol 2002;545:961-74.
- Shen YL, Chen YY, Wu XD, Bruce IC, Xia Q. Activation of mitochondrial ATP-sensitive potassium channels delays ischemia-induced cellular uncoupling in rat heart. Acta Pharmacol Sin 2004;25:22-8.
- Takashi E, Wang Y, Ashraf M. Activity of mitochondrial KATP channel elicits late preconditioning against myocardial infarction via protein kinase C signaling pathway. Circ Res 1999;85:1146-53.
- AN J. Varadarajan SG, Novalija E, Stowe DF. Ischemic and anesthetic preconditioning reduces cytosolic [Ca2+] and improves Ca2+ responses in intact hearts. Am J Physiol Heart Circ Physiol 2001;281:1508-23.
- Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-Xl. J Biol Chem 1999;274:2225-33.
- Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, et al. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA 1998;95:14681-6.
- Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev 2002;5:101-27.
- Wu C, Fujihara H, Yao J, Qi S, Li H, Shimoji K, et al. Different expression patterns of Bcl-2, Bcl-xl, and Bax proteins after sublethal forebrain ischemia in C57Black/Crj6 mouse striatum. Stroke 2003;34:1803.
- Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, et al. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA 2001;98:15318-23.