
Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats1
Introduction
Peripheral nerve injury induced in various ways may produce chronic pain states characterized by hyperalgesia, allodynia and spontaneous pain. To date, there is no effective treatment for releasing neuropathic pain. Recently, the synaptic plasticity of spinal cord neurons induced by long-lasting nociceptive stimulation (also called central sensitiza-tion) has been under intensive investigation[1–3]. Several studies have suggested that central sensitization related to pain, and hippocampal long term potentiation (LTP) associated with learning and memory, may share certain mechanisms[4,5]. For example, activation of N-methyl-D-aspartate (NMDA) receptor and the subsequent associated intracellular signal transduction cascades are involved in the induction, development and maintenance of synaptic plasticity in the spinal cord and hippocampus. Like the consolidation of early-phase LTP into late-LTP in the hippocampus, activity-dependent gene expression or transcription, which can increase the expression of pain-related receptors and signal proteins, plays an important role in conversion from acute nociceptive injury to chronic pain states[6,7]. The transcription factor cAMP response element binding protein (CREB), which can be phosphorylated by multiple intracellular kinases in response to a vast range of physiological and pathological stimuli, is critical for activity-dependent gene expression. Genetic studies have shown that CREB contributes to hippocampal late-LTP and memory consolida-tion, and CREB activation may serve as a molecular switch to transform short-lasting into long-lasting synaptic plasticity in the hippocampus[8,9]. Similarly, CREB has been suggested to contribute to the central sensitization associated with persistent pain states[10–12]. It has been proposed that NMDA activation-induced Ca2+ influx can trigger an early phase of CREB phosphorylation and a persistent phase of CREB phosphorylation is mediated by a delayed extracellular signal-regulated kinase (ERK) signal cascade, which is important to the development and maintainment of chronic pain.
ERK, one member of the mitogen-activated protein kinase (MAPK) family, transduces a broad range of extracellular stimuli into diverse intracellular responses by producing changes in the level of gene expression or transcription. Activated ERK translocates from the cytosol into the nucleus and in turn phosphorylates CREB at serine residue 133. Phosphorylation of CREB binding to the cAMP response element (CRE) of the target gene regulates gene expression and mediates the roles of ERK. Many studies found that ERK-mediated CREB phosphorylation is required for the induction of stable, late-phase LTP and long-term memory[13–15]. It has been reported that ERK may be involved in the modulation of nociceptive information and central sensitization produced by intense noxious stimuli and/or peripheral tissue inflammation[16–21]. However, few studies have focused on the roles of ERK and the relationship between ERK and CREB in neuropathic pain produced by nerve injury, such as chronic constriction injury (CCI) of the sciatic nerve.
In the present study, we used a CCI model to investigate whether activation and translocation of ERK were involved in the induction and maintenance of chronic neuropathic pain, and to observe the effect of ERK on the expression of pCREB in chronic neuropathic pain.
Materials and methods
Animals One hundred and seventy-four male Sprague-Dawley rats (200–250 g) provided by the Experimental Animal Center of Xuzhou Medical College were kept under a 12 h/12 h light-dark cycle regimen, with free access to food and water. All experiments were approved by the Animal Care and Use Committee at the Xuzhou Medical College and were in accordance with the college’s guidelines for the care and use of laboratory animals.
Implantation of intrathecal catheter For intrathecal drug administration, rats were implanted with catheters as described by Yaksh and Rudy[22]. In brief, under anesthesia with pentobarbital sodium (40 mg/kg, ip), rats were fixed, the occipital muscles were bluntly separated, then the cisternal membrane was exposed. Polyethylene catheters (PE-10) were
Chronic constrictive injury CCI of the sciatic nerve was performed as previously described by Bennett and Xie[23]. Briefly, under anesthesia with isoflurane, the left sciatic nerves of rats were exposed at the level of the middle of the thigh, and then four ligatures (4–0, silk thread) were tied loosely around proximal to the sciatica’s trifurcation at 1.0-mm intervals. Sham surgery was done by exposing the left sciatic nerve without ligation.
Drug administration Intrathecal drug administration was accomplished using a microinjection syringe connected to the intrathecal catheter in awake, briefly restrained rats. The injection was performed manually over a 30 s period in a single injection volume of 10 µL followed by a flush with 10 µL physiological saline.
U0126 (Biomol Research Laboratories, Pennsylvania, USA), an MEK inhibitor [dissolved in 5% dimethylsulfoxide (Me2SO), 0.5 g/L] or ODN (1 g/L) were administered every 12 h, 1 d pre-CCI and 3 d post-CCI. Me2SO and mis-sense ODN were injected as control.
The sequence of ERK antisense ODN was designed as reported previously[24]; antisense: 5'-GCCGCCGCCGCC-GCCAT-3', directed against the initiation of the translation start site of rat ERK1 and ERK2 mRNA; mis-sense: 5'-CGCGCGCTCGCGCACCC-3'. ODN was synthesized by Shanghai Sangon (Shanghai, China) and modified with phosphorothioate. The efficacy of the antisense oligonucleotide in inhibiting ERK expression was confirmed at the protein level by Western blotting. Twelve hours after the last administration of the antisense oligonucleotide, the expression of the ERK protein was reduced by approximately 70% in the rat spinal cord (Figure 1).

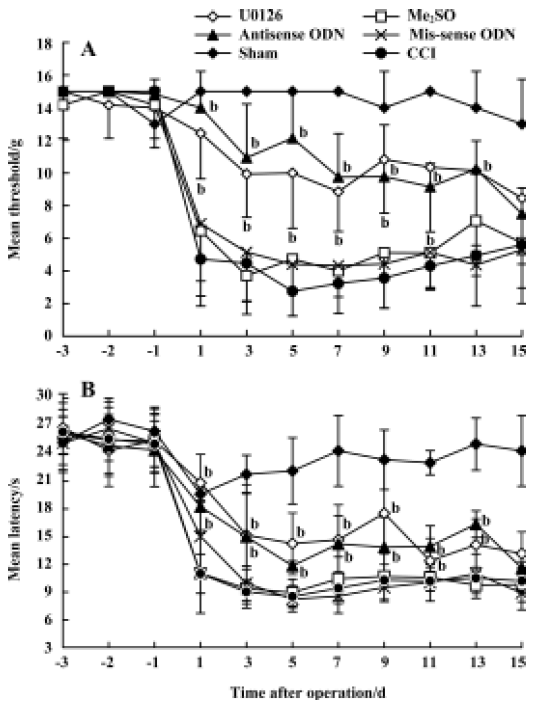
Behavioral studies Mechanical allodynia was assessed by using von Frey filaments. Animals were placed on a wire mesh platform and covered with a transparent plastic dome (20 cm×25 cm×15 cm), and the animals were allowed to acclimate to their surrounds for 30 min before testing. Each filament was applied perpendicularly to the plantar surface of the hindpaw (ipsilateral to the side of surgery in nerve-injured animals). The paw withdrawal threshold (PWT) was determined by sequentially increasing and decreasing the stimulus strength (the “up- and-down” method), and the data were analyzed using the nonparametric method of Dixon, as described by Chaplan et al[25].
Thermal hyperalgesia was assessed with the paw withdrawal latency (PWL) to radiant heat according to the protocol of Hargreaves et al[26]. Rats were placed in clear plastic cages on an elevated glass plate and allowed to acclimate to their surrounds for 30 min before testing. A high intensity light beam was focused onto the plantar surface of the hindpaw through the glass plate. The nociceptive endpoints in the radiant heat test were the characteristic lifting or licking
The tests were performed on each of 3 successive days prior to surgery and alternate days up to 15 d after surgery. Eight animals per group were used for behavior tests.
To study the effect of U0126 and antisense-ODN on the rat’s motor function, motor functions were evaluated by the observation of placing/stepping reflexes and righting reflexes and were conducted 5 min before the assessment of nociceptive responses.
Immunohistochemistry Immunohistochemical studies were performed as previously described[27,28]. Briefly, the spinal cord was cut into 40 µm-thick segments. After washing in phosphate buffer saline, the tissue sections were incubated in phosphate-buffered saline containing 5% normal goat serum and 0.3% TritonX-100 at room temperature for 30 min, followed by primary polyclonal mouse-anti-pERK antibody (1:200), primary polyclonal rabbit-anti-ser133-pCREB (1:400) antibody or primary polyclonal rabbit-anti-Fos antibody (1:1000) at 4 °C for 48 h (all antibodies were from Santa Cruz Biotechnology, California, USA). The sections were then incubated in biotinylated goat anti-rabbit or -mouse IgG (1:200) at 37 °C for 1 h and in avidin-biotin-peroxidase complex (1:100) (Vector Labs, California, USA) at 37 °C
We selected 5 spinal cord sections per animal, selecting sections that had the greatest number of positive neurons. For each animal, two measurements were made: (1) total number of positive neurons in the spinal cord dorsal horn ipsilateral to injury; (2) total number of positive neurons in the spinal cord dorsal horn contralateral to injury. All positive neurons were counted without considering the intensity of the staining.
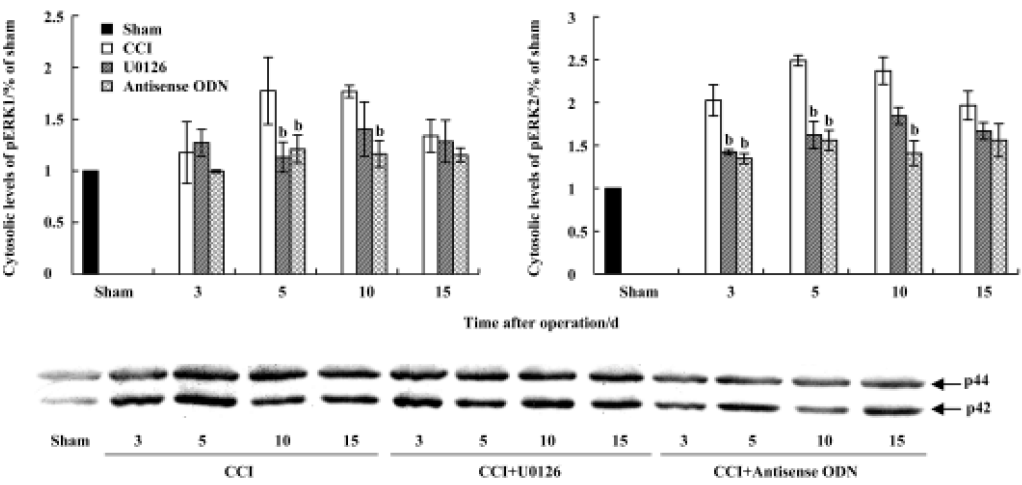
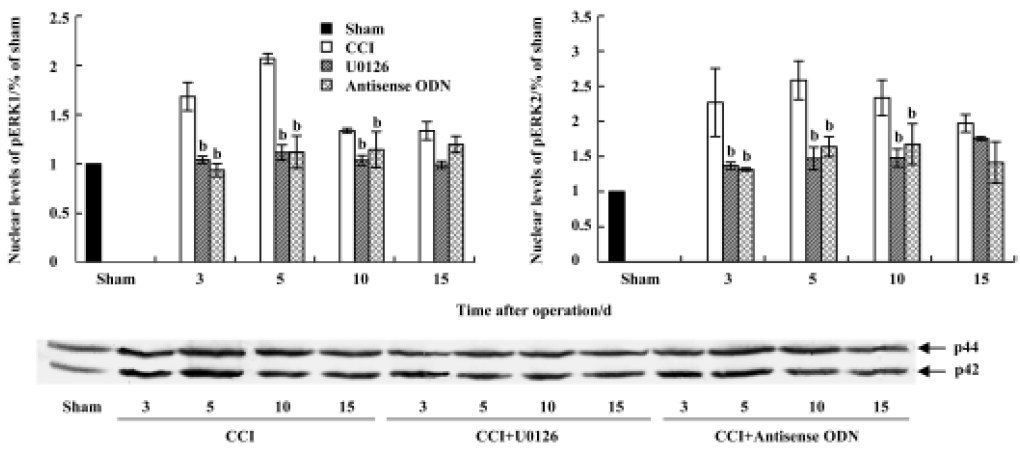
Western blotting The lumbosacral spinal cords of the rats were extracted and stored in liquid nitrogen. Tissue samples were homogenized in lysis buffer A (in mmol/L): HEPES 10.0, Na3VO4 1.0, MgCl2 1.5, KCl 10.0, NaF 50.0, edetic acid (EDTA) 0.1, egtazic acid (EGTA) 0.1, phenylmethylsul-fonyl fluoride (PMSF) 0.5, dithiothreitol (DDT) 1.0 and 0.02% protease inhibitor cocktail (pH 7.9). After the addition of 90 μL NP-40 (10%), the homogenates were vortexed for 30 s and then centrifuged at 800×g for 15 min at 4 °C. The supernatants were used for Western blot analysis as cytosolic proteins. The nuclear pellets were resuspended in buffer B (in mmol/L): HEPES 20.0, NaCl 420.0, MgCl2 1.5, EDTA 1.0, EGTA 1.0, PMSF 0.5, DDT 1.0, 20% glycerol, and 0.02% protease inhibitor cocktail (pH 7.9). The homogenates were incubated for 30 min in ice-cold water with constant agitation and then centrifuged at 13 000×g for 15 min at 4 °C to separate the nuclear proteins. Protein concentrations were determined using the Bradford method and the protein samples were stored at -80 °C.
Protein samples were dissolved in 4×sample buffer [in mmol/L: Tris-HCl 250.0, sucrose 200.0, DDT 300.0, 0.01% Coomassie brilliant blue-G, and 8% sodium dodecyl sulfate (SDS), pH 6.8], and denatured at 95 °C for 5 min, then the equivalent amounts of proteins were separated by using 10% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto a nitrocellulose membrane. The membranes were incubated overnight at 4 °C with the following primary antibodies: mouse polyclonal anti-pERK antibody and rabbit polyclonal anti-ser133-pCREB. The membranes were extensively washed with Tris-Buffered Saline Tween-20 (TBST) and incubated for 1 h with the secondary antibody conjugated with alkaline phosphatase (AP) at room temperature. The immune complexes were detected by using a NBT/BCIP assay kit (Promega, Shanghai, China). The scanned images were imported into Adobe Photoshop software (Adobe, California, USA). Scanning densitometry was used for semiquantitative analysis of the data.
Experimental groups Rats were divided into six groups in the behavior test: the sham group, CCI group, U0126 group, Me2SO group, antisense ODN group and mis-sense ODN group. Four groups were included in the immunohistochemistry studies and the Western blot testing: the sham group, CCI group, U0126 group, and antisense ODN group. Because few pERK, pCREB and Fos-positive neurons were expressed in the spinal dorsal horn of sham group rats, these data are not shown in results.
Statistical analysis All data are expressed as mean±SD Statistical analysis was carried out using one-way ANOVA or Student’s t-test. P<0.05 was considered statistically significant.
Results
Effects of U0126 or ERK antisense ODN on CCI-induced mechanical allodynia and thermal hyperalgesia Intrathecal administration of U0126 or ERK antisense-ODN did not affect the mechanical PWT, the thermal PWL, or motor function in the rats that received implanted intrathecal catheters. CCI, and not sham surgery, produced significant mechanical allodynia and thermal hyperalgesia. The time course of PWT and PWL is presented in Figure 2. Intrathecal injection of U0126 or ERK antisense ODN, and not Me2SO or mis-sense ODN, attenuated CCI-induced mechanical allodynia and thermal hyperalgesia.
Effects of U0126 or ERK antisense ODN on CCI-induced pERK expression in the spinal cord CCI significantly increased the expression of pERK-IR neurons in the spinal cord dorsal horn ipsilateral to injury. The pERK positive neurons were distributed mainly in laminae I and II of the spinal dorsal horn. Few positive neurons were distributed in other laminae or the contralateral spinal cord. Intrathecal injection of U0126 or ERK antisense ODN inhibited the increase in pERK expression in the spinal dorsal horn on postoperative d 3 and d 5 (Figure 3).

CCI-induced ERK activation was also confirmed by Western blot analysis. Compared with sham group rats, the levels of both phospho-ERK1 (44 kDa) and phospho-ERK2 (42 kDa), not unphospho-ERK (data not shown), were increased at all measured time points after CCI (Figures 4, 5). The expression of pERK reached a peak level on postoperative d 5. The increase in the nuclear fraction of pERK expression indicated that pERK was translocated into the nucleus from the cytoplasm. Intrathecal injection of U0126 or ERK antisense ODN markedly inhibited the increase in both cytosolic and nuclear pERK expression.
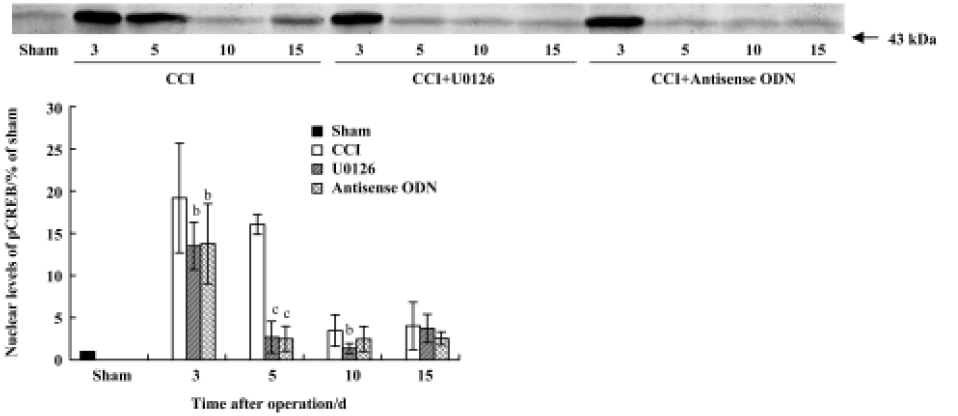
Effects of U0126 or ERK antisense ODN on CCI-induced pCREB expression in the spinal cord The immunohistochemical results revealed that CCI significantly increased the expression of pCREB, and pCREB-positive neurons were distributed in all laminae of the bilateral spinal cord. Expressions in the ipsilateral and contralateral spinal cord were not significantly different. Intrathecal injection of U0126 or ERK antisense ODN markedly inhibited pCREB expression in both sides of the spinal cord (Figure 6). Few pCREB positive neurons were found in the sham group rats (data not shown).
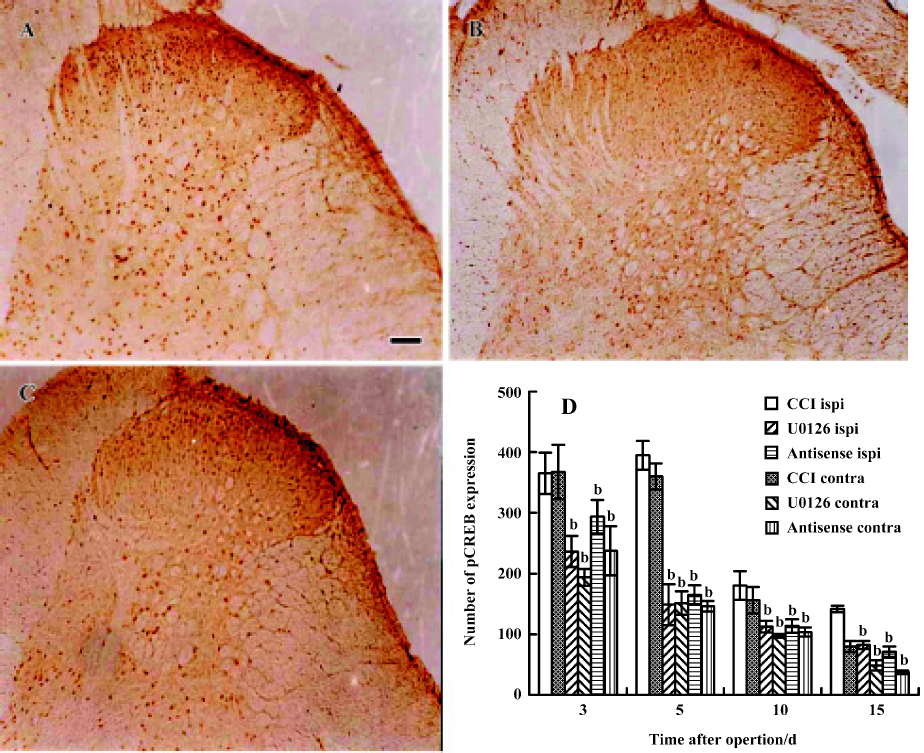
Similar to the results of the immunohistochemical studies, the Western blot results showed that nuclear pCREB was highly expressed on postoperative d 3 and d 5. Intrathecal administration of U0126 or ERK antisense ODN significantly inhibited the increase of pCREB expression, especially on postoperative d 5 (Figure 7). These data suggest that activation of ERK contributes to increased pCREB expression in the spinal cords of CCI rats.
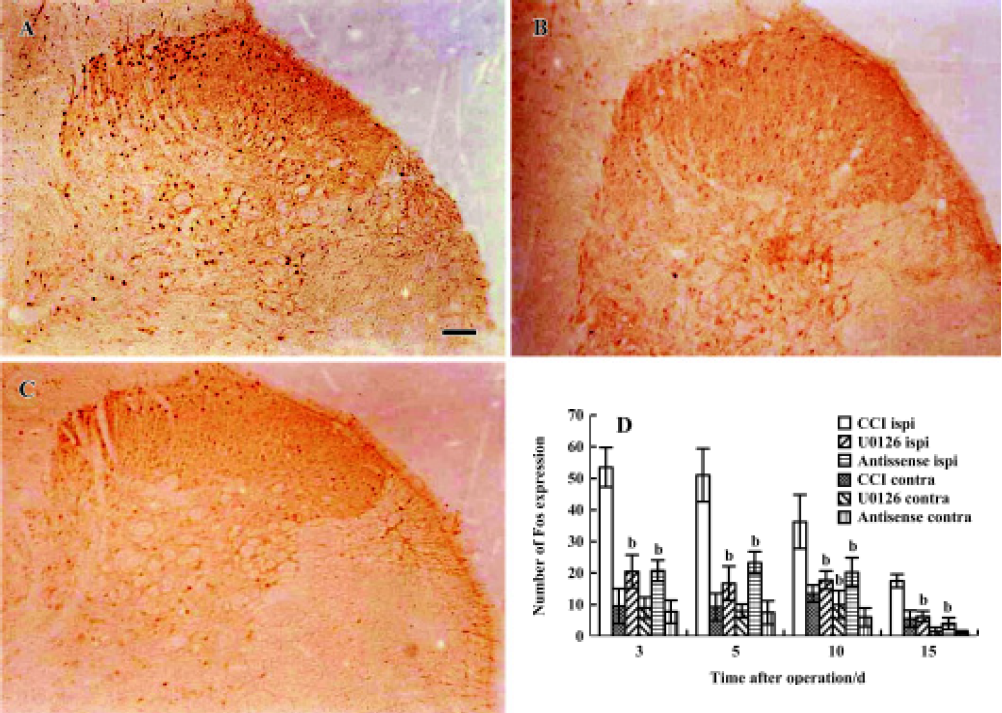
Effects of U0126 or ERK antisense ODN on CCI-induced Fos expression in the spinal cord CCI significantly increased the expression of Fos in laminae I and II of the ipsilateral spinal dorsal horn. Dissimilar to the findings for pCREB, few c-Fos positive neurons were found in the contralateral spinal cord. Intrathecal administration of U0126 or ERK antisense ODN also significantly reduced the expression of Fos-positive neurons (Figure 8A, 8B, 8C, and 8D).
Discussion
Central sensitization, an activity-dependent functional plasticity, is one of the main causes of behavior hyperalgesia under pathological conditions. Activation of postsynaptic membrane receptors or ion channels, intracellular kinase cascades and intranuclear gene expression contributes to the induction, development and maintainment of central sensitization. Intracellular kinase cascades, as the linkage bridge, transduce noxious stimuli into diverse intracellular responses, including changes in the levels of gene expression or transcription. A large number of studies have implicated several protein kinases, for example, protein kinases A (PKA), protein kinases C (PKC) and calcium/calmodullin-dependent protein kinase II (CaMKII), in central sensitization[29–31]. Recently, several studies have reported that ERK, a mitogen-activated protein kinase, contributes to pain hypersensitivity and central sensitization. Acute noxious stimuli, such as formalin or capsaicin, induce ERK phosphorylation in spinal dorsal horn neurons, and MEK inhibitor PD 98059 or U0126 reduces acute pain behavior after subcutaneous injection of formalin or capsaicin[16–21]. In agreement with previous reports using other pain models, the present studies indicate that activation of ERK in rat spinal cord contributes to CCI-induced allodynia and hyperalgesia. We demonstrated here, the time course of pERK and the relationship between pERK and pCREB expression in a CCI model. CCI induces long-lasting ERK phosphorylation and nuclear translocation in the spinal cord. The time course of changes in pERK expression, at least in part, correlated significantly with behavior hyperalgesia. Intrathecal injection of U0126 or ERK antisense ODN markedly attenuated CCI-induced mechanical allodynia and thermal hyperalgesia.
Nerve fibers display abnormal ectopic excitability at or near the site of nerve ligation after CCI. The local persistent abnormal excitation of sensory nerves can spread to the peripheral nerve bodies in the dorsal root ganglion (DRG) and central nervous system. Repeated or prolonged noxious stimulation and the persistent abnormal input following nerve injury increase the release of nociceptive neurotransmitters, such as glutamate, substance P, and calcitonin gene-related peptide (CGRP) in the central terminals of primary sensory afferents and then activate NMDA and neurokinin (NK) receptors in spinal dorsal horn neurons. Calcium influx through NMDA receptors triggers an increase in the levels of Ras-GTP and in turn leads to the activation of raf/MEK/ERK cascades.
ERK mediates central sensitization by phosphorylating the effector protein. Two potential effectors of ERK related to central sensitization are the potassium channel Kv4.2 and the transcription factor CREB. Recent evidence has shown that ERK integrates PKA and PKC to modulate the K+ channel Kv4.2 and A-type K+ currents (IA) in superficial dorsal horn neurons. Kv4.2 and IA are critical determinants of neuronal excitability in central sensitization[32–34]. It is possible that modulation of potassium channels may be involved in the short-term or acute effect of pERK. Our unpublished data show that CCI-induced hyperalgesia is reversed 30 min after intrathecal administration of U0126 on postoperative d 5.
Importantly, pERK is able to translocate from the cytoplasm into the nucleus and in turn phosphorylate the transcriptional factor CREB on serine 133. In the current study, CCI significantly increased the expression of pCREB in the rat spinal cord. The time course of pCREB expression correlates with the activation of ERK and behavioral hyperalgesia. Intrathecal injection of U0126 or ERK antisense ODN markedly inhibited the increase in pCREB expression. This suggests that activation of ERK may contribute to increased pCREB expression in the spinal cord of CCI rats, and that the function of pERK is partly accomplished via CREB-dependent gene expression. Phosphorylation of CREB on serine 133 recruits the CREB binding protein, CBP, to the complex and promotes the transcription of downstream genes. Many “pain genes”, which may contribute to central sensitization, are activated by CREB, including the immediate early gene c-fos, BDNF, CGRP, the alpha subunit of CaMKII, and the neurokinin 1 receptor. A considerable amount of evidence has indicated that CREB-dependent gene expression is required for long-term changes in the synaptic plasticity induced by various nociceptive stimuli[10–12,35–37].
We found that increases in pCREB expression occurred bilaterally in all laminae of the spinal cord in CCI rats. Expression levels in the ipsilateral and the contralateral sides were not significantly different. Fos protein was expressed only in the ispilateral spinal dorsal horn. Similar results have been reported in other pain models, for example, formalin injection and carrageenan injection[11,12,38,39]. However, few interpreted this result consistently. Ji and Rupp speculated that signal strength might affect gene expression differentially with respect to protein phosphorylation[38]. We, however, favor the explanation that different intracellular signal transduction pathways contribute to pCREB expression in the ispilateral and the contralateral spinal cords. In the present study, pCREB was expressed in both sides of the spinal cord, but pERK only in the ipsilateral side. These results suggest that pERK contributed to pCREB expression in the ipsilateral spinal cord, but not in the contralateral spinal cord. Intrathecal injection of U0126 or ERK antisense ODN inhibited pCREB expression not only in the ispilateral spinal cord, but also in the contralateral spinal cord. This suggests that activation of ERK in the ispilateral spinal cord is also involved in pCREB expression in the contralateral spinal cord. CCI induces ipsilateral primary afferent terminals to release nociceptive neurotransmitters, which induces the NMDA receptor-dependent activation of intracellular kinase cascades, including the ERK pathway, in the spinal cord. Furthermore, cross-talk between intracellular kinase cascades can produce strong activation of signal pathways. Then some kinases phosphorylate CREB and regulate noxious-induced target gene expression, such as c-fos, in a pCREB-dependent manner. But how should we interpret pCREB expression in the contralateral spinal cord, which did not receive noxious inputs? Numerous lines of evidence have shown that unilateral noxious stimuli can indirectly induce the activation of several intracellular signal pathways in the bilateral spinal cord. For example, Solodkin et al reported that unilateral hindpaw inflammation produced a bilateral increase in NADPH-diaphorase in rat lumbar spinal cord[40]. Noxious stimuli also induce glial activation, and excitation of glia at one site can activate distant and even the contralateral dorsal horn glia via gap junctions and propagated calcium waves[41]. The ipsilateral noxious stimuli-induced activation of several intracellular signal pathways in the contralateral spinal cord may indeed be a critical factor in pCREB expression in the contralateral spinal cord. Because CREB phosphorylation in the contralateral spinal cord does not result from direct extracellular noxious inputs, it cannot upregulate the expression of the noxious marker gene, c-fos.
In summary, the present study found that activation of the ERK/CREB pathway in the spinal cord contributed to chronic constrictive injury-induced neuropathic pain.
Acknowledgements
We gratefully acknowledge technical assistance from Dr Hai-lei DING. We also thank Steve PILLING for editing the English of this manuscript.
References
- Ikeda H, Heinke B, Ruscheweyh R, Sandkuhler J. Synaptic plasticity in spinal lamina. I projection neurons that mediate hypera-lgesia. Science 2003;299:1237-40.
- Melzack R, Coderre TJ, Katz J, Vaccarino AL. Central neuroplas-ticity and pathological pain. Ann NY Acad Sci 2001;933:157-74.
- Wolpaw JR, Tennissen AM. Activity-dependent spinal cord plasticity in health and disease. Annu Rev Neurosci 2001;24:807-43.
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 2003;26:696-705.
- Rygh LJ, Tjolsen A, Hole K, Svendsen F. Cellular memory in spinal nociceptive circuitry. Scand J Psychol 2002;43:153-9.
- Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR. Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci 2002;22:7453-61.
- Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA 1999;96:7723-30.
- Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, et al. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron 2003;39:655-69.
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, et al. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 2000;20:6459-72.
- Hoeger-Bement MK, Sluka KA. Phosphorylation of CREB and mechanical hyperalgesia is reversed by blockade of the cAMP pathway in a time-dependent manner after repeated intramuscular acid injections. J Neurosci 2003;23:5437-45.
- Ma W, Hatzis C, Eisenach JC. Intrathecal injection of cAMP response element binding protein (CREB) antisense oligonucleotide attenuates tactile allodynia caused by partial sciatic nerve ligation. Brain Res 2003;988:97-104.
- Ma W, Quirion R. Increased phosphorylation of cyclic AMP response element-binding protein (CREB) in the superficial dorsal horn neurons following partial sciatic nerve ligation. Pain 2001;93:295-301.
- Kelleher RJ, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 2004;116:467-79.
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, et al. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 2003;39:309-25.
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nature Rev Neurosci 2004;5:173-83.
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci 2001;21:6933-9.
- Ciruela A, Dixon AK, Bramwell S, Gonzalez MI, Pinnock RD, Lee K. Identification of MEK1 as a novel target for the treatment of neuropathic pain. Br J Pharmacol 2003;138:751-6.
- Galan A, Cervero F, Laird JM. Extracellular signaling-regulated kinase-1/2 (ERK1/2) mediate referred hyperalgesia in a murine model of visceral pain. Mol Brain Res 2003;116:126-34.
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensi-tivity. Nat Neurosci 1999;2:1114-9.
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci 2002;22:478-85.
- Wang H, Dai Y, Fukuoka T, Yamanaka H, Obata K, Tokunaga A, et al. Enhancement of stimulation-induced ERK activation in the spinal dorsal horn and gracile nucleus neurons in rats with peripheral nerve injury. Eur J Neurosci 2004;19:884-90.
- Yaksh TL, Rudy TA. Chronic catheterization of the subarachnoid space. Physiol Behav 1976;7:1032-6.
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorder of pain sensation like those seen in man. Pain 1988;33:87-107.
- Sale EM, Atkinson PG, Sale GJ. The requirement of MAP kinase for the differentiation of fibroblasts to adipocytes, for insulin activation of p90 S6 kinase and for insulin or serum stimulation of DNA synthesis. J EMBO 1995;14:674-84.
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TJ. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55-63.
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988;32:77-88.
- Cao JL, Zeng YM, Zhang LC, Gu J, Liu HF, Zhou WH, et al. NO mediated the increase of Fos protein and NDMA1AR mRNA expression in the rat spinal cord during morphine withdrawal. Acta Pharmacol Sin 2001;22:505-11.
- Cao JL, Ding HL, Zhang LC, Duan SM, Zeng YM. Pretreatment with midazolam suppresses morphine withdrawal response in mice and rats. Acta Pharmacol Sin 2002;23:685-90.
- Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 1999;19:2181-6.
- Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci 2002;22:4196-204.
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol 2001;429:23-37.
- Hu HJ, Glauner KS, Gereau RW IV. ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I. Modulation of A-type K+ currents. J Neurophysiol 2003;90:1671-9.
- Hu HJ, Gereau RW IV. ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol 2003;90:1680-8.
- Adams JP, Anderson AE, Varga AW, Deneley KT, Cook RG, Pfaffinger PJ, et al. The A-type potassium channel KV4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem 2000;75:2277-87.
- Lewin MR, Walters ET. Cyclic GMP pathway is critical for inducing long-term sensitization of nociceptive sensory neurons. Nat Neurosci 1999;2:18-23.
- Miletic G, Pankratz MT, Miletic V. Increases in the phosphorylation of cyclic AMP response element binding protein (CREB) and decreases in the content of calcineurin accompany thermal hyperalgesia following chronic constriction injury in rats. Pain 2002;99:493-500.
- White DM, Walker S, Brenneman DE, Gozes I. CREB contributes to the increased neurite outgrowth of sensory neurons induced by vasoactive intestinal polypeptide and activity-dependent neurotrophic factor. Brain Res 2000;868:31-8.
- Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci 1997;17:1776-85.
- Messersmith DJ, Kim DJ, Iadarola MJ. Transcription factor regulation of prodynorphin gene expression following rat hindpaw inflammation. Mol Brain Res 1998;53:260-9.
- Solodkin A, Traub RJ, Gebhart GF. Unilateral hindpaw inflammation produces a bilateral increase in NADPH-diaphorase histochemical staining in the rat lumbar spinal cord. Neuroscience 1992;51:495-9.
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci 2003;23:1026-40.