
Preparation of liposomal brucine and its pharmaceutical/pharmacodynamic characterization1
Introduction
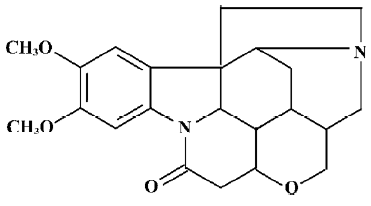
Brucine is an alkaloid and exists mainly in the seeds of Strychnos nux-vomica L (Loganiaceae)[1], which is widely used in many southern Asian countries. The configuration of brucine is shown in Figure 1.
Brucine itself is known as an anti-inflammatory and analgesic drug for relieving arthritic and traumatic pain[2]. Its main pharmacodynamic actions include relief of pain, reduction of swelling, and the promotion of circulation. Unfortunately, the potential use of brucine is severely limited due to its high incidence of side-effects, including violent convulsion and even lethal poisoning[3]. Therefore, for a therapeutic application of this molecule, it is necessary to find a suitable formulation to limit its dangerous side-effects while maintaining or possibly enhancing its effectiveness.
Colloidal drug delivery systems, such as liposomes represent a mature, versatile technology with considerable potential for the entrapment of both lipophilic and hydrophilic drugs[4]. Encapsulation or entrapment of drugs in liposomes results in distinct changes of pharmacokinetic and pharmacodynamic properties of free drugs, and in some cases, causes an apparent decrease in toxicity and/or an increase in therapeutic efficacy[5–7]. Meanwhile, liposomes have also been suggested to be useful carriers for topical and trans-dermal delivery[8,9].
Therefore, the objectives of our study are: (i) to develop a safe and effective liposomal brucine (LB) which achieves attenuation and synergia of brucine; (ii) to achieve topical and transdermal administration of the drug; (iii) to assess the acute dermal toxicity and skin irritation; and (iv) to evaluate the analgesic and anti-inflammatory activities of LB using acetic acid-induced writhing test and xylene-induced mouse ear edema models.
Materials and methods
Materials Brucine was purchased from Fluka (Buchs SG, Switzerland). Lecithin (from soy beans, ≥92%), cholesterol, sodium deoxycholate, and mannitol were obtained from Yuanju Biotech (Shanghai, China). Yunnan Baiyao tincture was purchased from Yunnan Baiyao Group (Kunming, China), and xylene was obtained from Shanghai Chemical Reagent (Shanghai, China). Acetic acid and Tween-80 were from Sinopharm Chemical Reagent (Shanghai, China). All other reagents were of analytical grade.
Animals New Zealand albino rabbits of both sexes weighing 2 kg, nude mice (BALB/c) of both sexes weighing 18–22 g, and ICR (Kunming) mice of both sexes weighing 18–22 g were used for the experiments. The animals were housed in standard cages, maintained at a temperature of 22±2 °C with 12 h light/dark cycles, and provided with fresh water and standardized diets ad libitum, unless otherwise noted. The animal tests were performed according to the Principles of Laboratory Animal Care and Use in Research (Ministry of Health, Beijing, China).
Statistical analysis The results were expressed as mean± SD. All data were processed with SPSS software (SPSS, Chicago, IL, USA). P<0.01 was considered significant difference.
Preparation of liposomes A modified ethanol-dripping method was used to prepare LB[10]. An ethanol solution of lipid (lecithin: cholesterol =6:1, w/w), sodium deoxycholate, Tween-80, and brucine (16:4:4:1, w/w) was dripped into mannitol solution (5.3 mg/mL). The ratio of the ethanolic-lipid phase to the aqueous phase was 1:9 (v/v) in the final suspen-sion. The suspension was then sonicated (45 w, Shanghai Kedao Ultrasonic Instrument Co, Shanghai, China) with an ice water-bath for 20 min and freeze-dried for 72 h. The dry powder was stored at -18 °C in a refrigerator. The dry powder could be rehydrated and sonicated for 3 min prior to application.
Characterization of liposomes Non-encapsulated brucine was separated from the liposomes by gel filtration (Sephadex G-25 column, Shanghai, China), and encapsulated efficiency (EE) was calculated as the following equation:
EE=(total drug-unencapsulated drug)/total drug×100%.
The average diameters of liposomes were determined by dynamic light scattering using a laser lightscattering instrument (Nicomp, 380/ZLS, Santa Barbara, USA). The samples were diluted in distilled water in order to avoid multiple scattering. This measurement was performed at 25±0.1 °C, at an angle of 90 degrees between laser and detector.
The morphology of the liposomes was studied with an atomic force microscope (AFM). One sample was diluted with distilled water. One drop of that liquid was dripped onto a sheet of mica and characterized after it was dried by AFM (Multimode, Veeco, Santa Barbara, USA).
In vitro release A volume of 2 mL of LB or free brucine (drug content: 1 mg/mL) was taken in a dialysis tube (Poly-carbonate, molecular cut-off, 3500) with both the ends tied. The dialysis bag was suspended in phosphate buffer solution (PBS, 100 mL, pH 7.4), maintained at 37 °C, and the PBS was kept stirring. Periodically, 2 mL of PBS sample was drawn, and then the absorbance was noted at 263 nm using the spectrophotometer (756MC, Shanghai Yukang Instrument Co, Shanghai, China). The volume of PBS was maintained at 100 mL throughout the experiment.
In vitro skin permeation The in vitro skin permeation study was based on a previously reported method[11]. The skin fragment used for the experimentation was excised from nude mice and stored at 4 °C 1 d before the experiment. Subcutaneous fat was removed before the experiment started. The skin samples were mounted on Franz diffusion cells (diffusion surface: 3.14 m2). The receiver compartments were filled with 15 mL of (PBS; pH 7.4), kept at 37 °C, and continuously agitated by a magnetic stirrer. The donor chamber was charged with 1 mL LB and free brucine (drug content: 1 mg/mL). At different periods of time (1, 2, 4, 6, 8, and 12 h), 0.5 mL samples were withdrawn from the receptor compart-ment, replaced with an equal volume of fresh medium, and HPLC was employed for the drug content. The HPLC apparatus was Agilent 1100 (Santa Clara, USA) with a wavelength detector. The detection wavelength was 260 nm. Analysis was performed with a mixed solution (55% methanol and 45% acetonitrile), with a flow rate of 1.0 mL/min on a reverse column (Hypersil ODS C18 column, Shanghai, China) at 30±0.1 °C.
After a 12 h permeation period, the residual formulation from each skin fragment was cleaned with distilled water. Each skin sample was weighed, placed in 2 mL ethanol solution (50%), and disintegrated by an ultrasonic processor. After agitation for 2 min and centrifugation for 5 min at 1370 (×g), the supernatant was removed and filled with 2 mL ethanol solution (50%). After repeating the extractive process 5 times, all supernatants were collected. Then, after filtration, the drug concentration was determined by HPLC. The drug accumulation of brucine in the skin was calculated by the following formula:
Drug accumulation in skin=(Drug content in skin)/(skin weight).
Acute dermal toxicity An acute dermal toxicity study in rabbits was conducted in accordance with the study of Isbrucker et al[12]. Rabbits of both sexes were divided into 2 groups: integrity skin group and broken skin group. Each group had 3 subgroups: brucine group, LB group, and negative control group (saline solution, 0.9% NaCl); each subgroup consisted of 4 rabbits. Two sides of each rabbit’s back were clipped with an electric clipper to expose an area of approximately 10% of the total body surface 24 h before the experiment. Only those animals without injury or irritation of the skin were used in the test. Signs of “#” were cut on the 2 depilated parts of each animal in the broken skin group. The vehicle and drugs were topically administered within 24 h. Then the test sites were gently cleaned of any residual test substance. The animals were observed for mortality, signs of gross toxicity, and behavioral changes after application at least once daily thereafter 7 days.
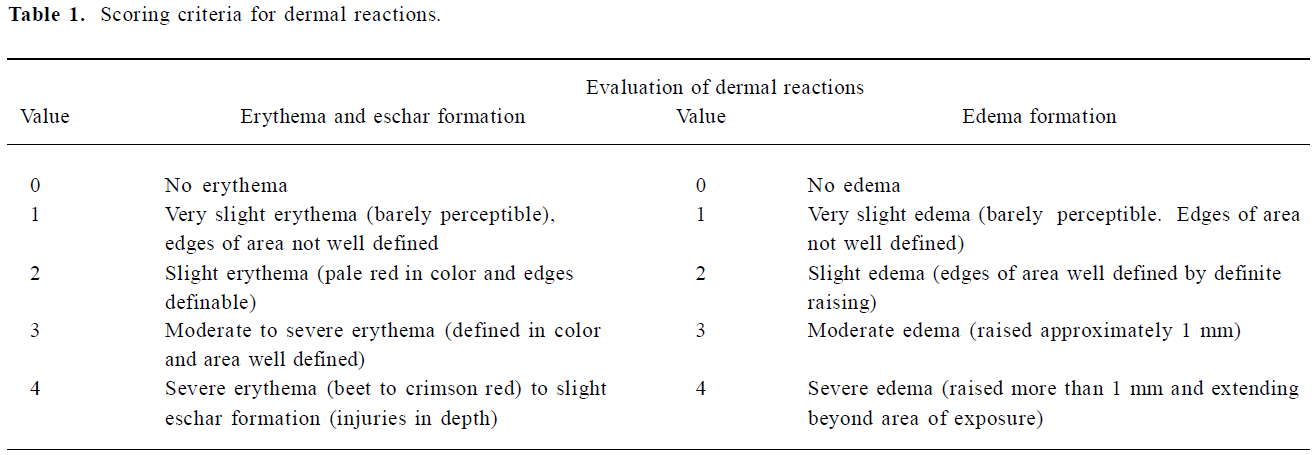
Skin irritation study A primary skin irritation test was conducted on the rabbits to determine the potential irritation reactivity according to a previous method[13]. The groups of rabbits were the same as those in the acute dermal toxicity test. Each animal was treated with vehicle and drug (1 mg/mL) which was applied to the skin of 1 flank. The patch was held in place with a bandage for 24 h, after which the dressing and patch were removed and the skin was cleaned of residual test substance with water. Skin reactions and irritation effects were assessed at approximately 1, 24, 48, and 72 h after the removal of the dressings. Adjacent areas of untreated skin from each animal served as controls. Erythema and edema were scored on a scale of 0–4, with 0 showing no effect and 4 representing severe erythema or edema (Table 1). The specific degree of irritancy was obtained by calculating the primary dermal irritation index (PDII, PDII=Σ(value of evaluation n of dermal reactions)/4) and classified according to the descriptive rating for mean primary dermal irritation index (0<PDII≤2.0: slight irritation; 2.0<PDII≤5.0: moderate irritation; and PDII >5.0: severe irritation).
Full table
Xylene-induced mouse ear edema The assay of xylene-induced ear edema in mice was based on a previously reported method[14]. Male mice were divided into 8 groups and each group consisted of 10 mice. The positive control group received Yunnan Baiyao tincture (3 mg/kg), the negative group received saline solution (0.9% NaCl), 3 groups received free brucine (1.5, 3, and 6 mg/kg), and another 3 groups received LB (1.5, 3, and 6 mg/kg, brucine content), respectively. The vehicle and drugs were topically applied to both sides of the right ear of the mice. The left ear received only saline solution. Thirty minutes after the administration of vehicle and drugs, 50 µL xylene was applied to both surfaces of the right ear. After treatment for 30 min, the mice were killed by cervical dislocation, and a plug (9 mm diameter) was removed from each ear. The edematous response was measured as the weight difference between the 2 plugs. The anti-inflammatory activity was expressed as the percentage of the inhibition of edema (PIE). PIE was calculated using the following formula:
PIE=([control weight variation-treated weight variation]/control weight variation)×100%
Acetic acid-induced writhing test The response to the intraperitoneal injection of 0.9% acetic acid was induced by the method proposed by Koster et al[15]. The groups of mice were the same as those in the xylene-induced ear edema test. The abdomens of the mice were clipped with an electric clipper 24 h before the experiment. Only those animals without injury or irritation of the skin were used in the test. The vehicle and drugs were topically administered on the abdomens of mice 30 min before the intraperitoneal injection of 1% acetic acid (10 mg/kg). The number of abdominal writhes within 20 min after the acetic acid injection was counted and the percentage protection (PP) was calculated using the following formula: PP=([control mean-treated mean]/control mean)×100%
Duration of analgesic and anti-inflammatory activities The acetic acid-induced writhing test and xylene-induced ear edema in mice were also used to investigate the duration of analgesic and anti-inflammatory activities of free brucine and LB. Each test had 3 groups: free brucine group (3 mg/kg), LB group (3 mg/kg), and the negative control group (saline solution). In the writhing test, the mice received an intraperitoneal injection of 1% acetic acid at 30, 120, and 240 min after the administration of vehicle/drugs. In the xylene-induced mouse ear edema test, after treatment with vehicle/drugs for 30, 120, and 240 min, respectively, 50 µL xylene was applied to both surfaces of the right ear. PP and PIE were calculated according to the formulas defined in xylene-induced mouse ear edema and acetic acid-induced writhing test sections.
Results
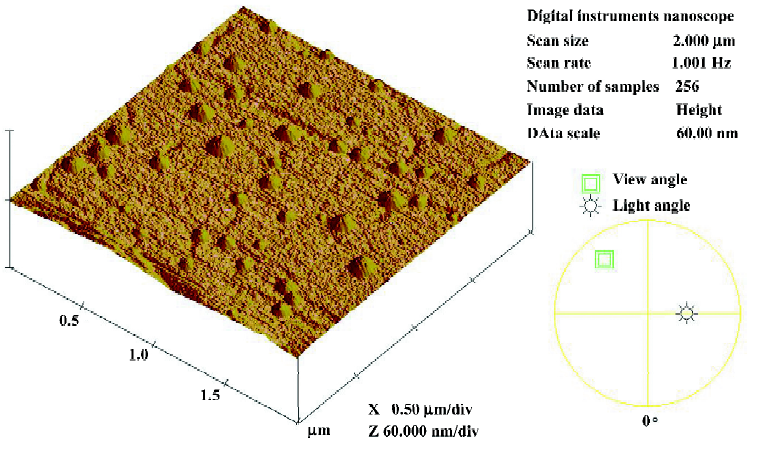
Characterization of liposomes The particle size distribution of the liposomes is shown in Figure 2. The average size of the liposomes was between 25 and 110 nm, and the mean diameter was 55.4 nm, indicating a narrow population distribution. The AFM image of liposomes is presented in Figure 3. It shows that the liposomes were discrete particles with round structures and sharp boundaries and ranged from 20 to 100 nm, identical to Figure 2. The EE of LB determined by gel filtration was 71.95%±2.65% (n=4).

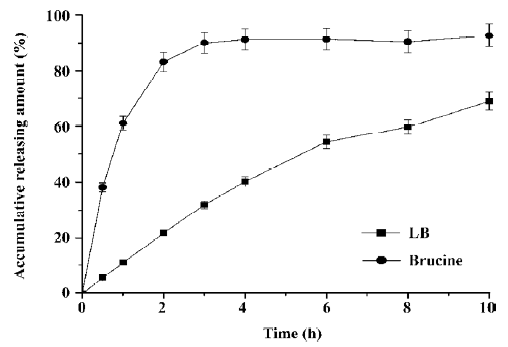
In vitro release and skin permeation studies The drug release profile of LB and free brucine through dialysis are listed in Figure 4. It could be observed from Figure 4 that before encapsulated, brucine diffused quickly and the diffusion amount was up to 90% in 2.5 h. But after encapsulated in liposome, the release rate of brucine was significantly decreased and only 68% of drug released into PBS buffer after 10 h. This result showed that the LB exhibited an obvious effect of slow release.
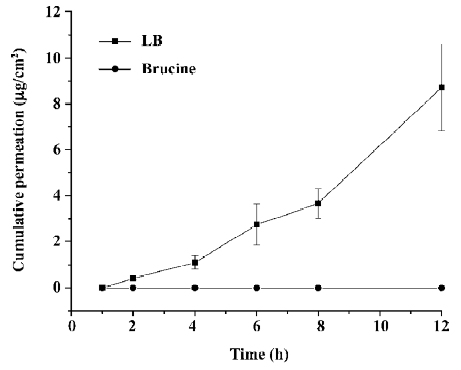
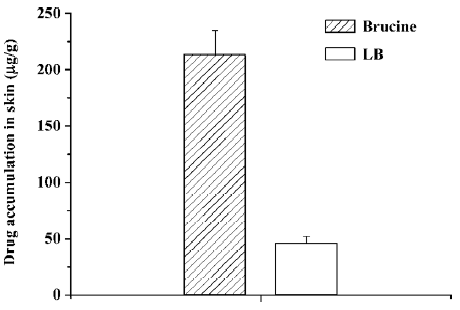
The amount of brucine permeation through skin is plotted in Figure 5. In this Figure, almost no detectable brucine was permeated through skin in the free brucine group in 12 h. But for the LB group, the cumulative drug permeated increased with the increasing of the time and reached 8.7 µg/cm2 after 12 h. The drug accumulation of LB and free brucine in skin is indicated in Figure 6. After 12 h permeation, only 46.8 µg/g brucine accumulated in the skin in the LB group. In the free brucine group, the accumulation reached 213 µg/g, which is over 5 times that of the LB group. These results showed that LB exhibited an obvious promotion of skin permeation.
Acute dermal toxicity The results of the acute dermal toxicity test are presented in Table 2. It can be found that as for the LB group and negative control group following the dermal application of LB (100 mg/kg body weight), the animals whether with integrity skin or broken skin, all survived, gained weight and appeared active and healthy. There were no signs of gross toxicity, dermal irritation, adverse pharmacologic effects or abnormal behaviors. But in the case of the free brucine groups, slight hyperaemia in integrity skin and hyperaemia and cyanosis in broken skin group were noted following the dermal application after removal of the dressing. Thereby the acute dermal toxicity LD50, to rabbits of both sexes was greater than 100 mg/kg. These results showed that the LB exhibited the attenuation of brucine.

Full table
Skin irritation study The objective of this study was to determine the potential of LB and brucine to produce irritation from a single topical application to the integrity and broken skin of New Zealand albino rabbits. The results of this experiment showed that after application for 1, 24, 48, and 72 h, no irritation was observed in the integrity group. While in the broken group, 24 h after the application of brucine, 1 case of erythema was observed and the severity of irritation increased with time. The specific PDII of the brucine subgroup in the broken skin group was 0.25, 0.5, and 1 after 24, 48, and 72 h, respectively. The results also showed the attenuation of brucine, which was similar to the results of the acute dermal toxicity study.
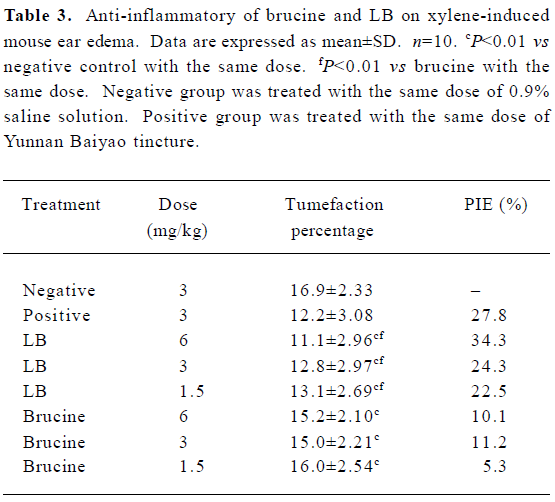
Xylene-induced mouse ear edema The anti-inflammatory effects of brucine and LB on xylene-induced mouse ear edema are presented in Table 3. Statistical analysis demonstrated that both LB and brucine showed significant anti-inflammatory effects (P<0.01). The maximal PIE of LB was 34.3% at the dose of 6 mg/kg, even exceeding that of the positive control (27.8%). Furthermore, LB, with doses ranging from 1.5 to 6 mg/kg, caused significant stronger inhibitory effects than the free brucine (P<0.01). The PIE of LB was 2–4 times that of free brucine in the same doses, and LB exerted its anti-inflammatory effects in a dose-dependent manner.
Full table
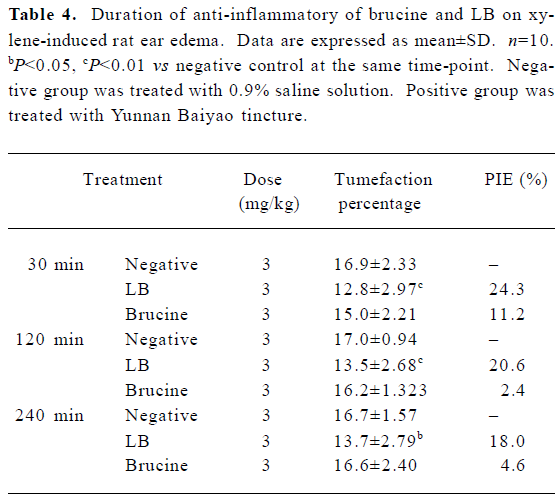
The results of the duration of brucine and LB on xylene-induced mouse ear edema are presented in Table 4. The results demonstrated that both brucine and LB exerted their maximal anti-inflammatory effects at 30 min after drug administration, 11.2% for brucine at a dose of 3 mg/kg, and 24.3% for LB. After 30 min, the anti-inflammatory effects of free brucine quickly decreased. At 120 and 240 min after administration, the PIE of free brucine were only 2.4% and 4.6%, while the PIE of the LB groups were 8 and 4 times that of free brucine, reaching 20.6% and 18.5%, respectively. This result demonstrated that LB has longer lasting anti-inflammatory effects than free brucine.
Full table
Acetic acid-induced writhing test The results of the dose-dependent response effects of brucine and LB on acetic acid-induced writhing in mice are presented in Table 5. It was observed that the LB and brucine groups significantly inhibited the writhing response of mice caused by the intraperitoneal administration of acetic acid with the exception of the free brucine at a dose of 1.5 mg/kg. The maximal inhabitation of the nociceptive response was 40.7% for LB at the dose of 6 mg/kg, which exceeded that of the positive control at 3 mg/kg (36.4%). Compared with the corresponding brucine groups at 1.5 and 6 mg/kg, the LB groups exhibited significantly better analgesic efficacy. At 6 mg/kg, the pain inhibition intensity of LB was 1.5 times higher than that of free brucine, which suggested that after being encapsulated, the analgesic efficacy of brucine was increased. Also, LB exerted its pain-relieving effect in a dose-dependent manner.
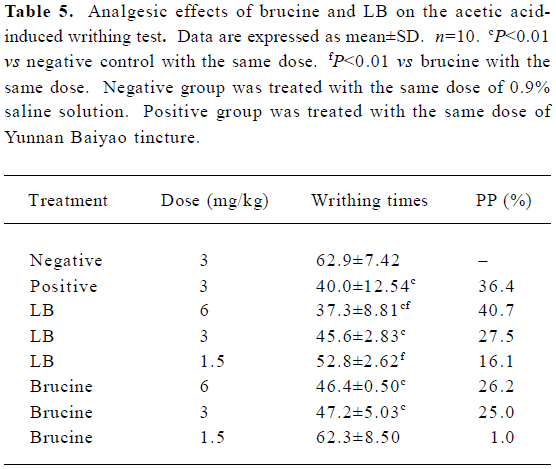
Full table
The results of the duration of the analgesic effects of brucine and LB are presented in Table 6. It showed that 30 min after drug administration, the analgesic effects of brucine and LB at the dose of 3 mg/kg all peaked, and the PP of brucine and LB were 25% and 27.5%, respectively. The PP of free brucine at 240 min after administration was 18.2%, while the PP of LB was still as high as 26.3%. The decrease of the analgesic effects of LB and brucine showed that LB exerted a more lasting analgesic effect.
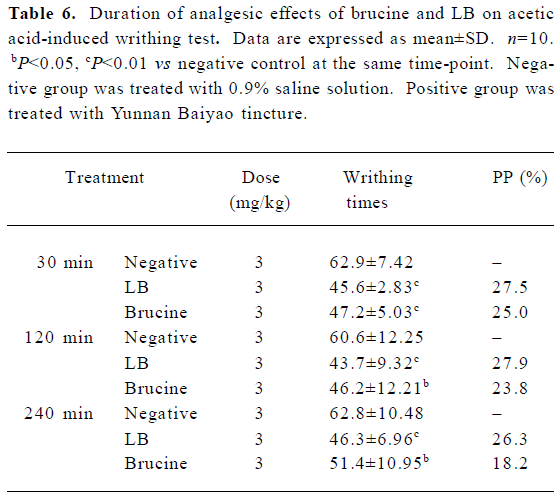
Full table
Discussion
Local application of drugs is promising as it allows a controlled and continuous delivery of drugs, and avoids or reduces systemic side-effects. The greatest limitation to the transdermal route is the skin barrier, which prevents the uptake of drugs. Our strategy to overcome the skin barrier is the encapsulation of drugs into liposomes, vesicles consisting of a lipid bilayer that act as carriers. Liposomes as drug delivery systems for drugs have great potential in providing controlled release, decreasing the toxicity of drug, and altering the bioavailability[5–7]. The following explanations will support this conclusion.
In the present study, a liposomal brucine formulation for transdermal delivery was prepared. The mean diameter of the prepared liposomes (Figure 2), ranging from 25 to 110 nm, was 55.4 nm, and the AFM micrograph (Figure 3) was further proof of the narrow size distribution of liposomes. The influence of the liposome size on the properties seemed to be contradictive. Verma and his co-workers reported that small liposomes were more suitable to penetrate skin than the large one with the same composition[16]. But small particles always led to low encapsulation efficiency, thereby decreasing the drug amount loaded. EE, standing for the efficiency of encapsulating brucine, in our study was 71.95%±2.65% (n=4), which was turned out to be a proper efficiency by the following in vitro and in vivo evaluation experiment. A high EE value was the foundation for achieving sustained-release, attenuation of toxic drugs and effectively transporting drugs.
In vitro release (Figure 4) exhibited an obviously sustained release of liposomes because only 68% brucine was released in 10 h in LB, while 90% of the drug was released in 2.5 h in the free brucine group. Our data for the duration of analgesic and anti-inflammatory activities (Tables 4, 6) also revealed that compared with the groups administrated by free brucine with the same dosage, the groups of LB exhibited 4–8 folds increase of PIE and significant higher PP after the same administration time, indicating that LB had longer pharmacodynamic action than that of the free brucine. Controlled/sustained release was a significant property of colloidal drug delivery systems. Our in vitro and in vivo studies demonstrated that liposomal formulation exerted an adequate controlled/sustained release, which led to long-lasting pharmacodynamic action and enhancing effects. These results were significant in improving bioavailability and decreasing the administering frequency of the drug.
It has been accepted that the most barrier of the trans-dermal administration route is the skin, especially the keratose layer, which greatly prevents the permeation and uptake of the drug. In vitro permeation test indicated that there was no detectable brucine permeation through the skin into receiver compartments and drug accumulation in the skin was 213 µg/g after 12 h for the free brucine group. In contrast, the cumulative drug permeation of the LB group reached 8.7 µg/cm2 while drug accumulation was just one-fifth of that of free brucine. Brucine, due to skin barrier and poor solubility of itself, scarcely penetrates through skin layers and might accumulate in skin. Liposomes, as useful carriers for topical drug delivery, can effectively improve the drug permeation through the skin, thereby decreasing the drug accumulation in the skin tissue. This mechanism is important and useful for some toxic drugs to reduce their dermal toxicity.
Because of the violent toxicity of brucine, the acute dermal toxicity and skin irritation of LB, as a dermal formulation, should certainly be investigated. Our study demonstrated the acute dermal LD50 of prepared liposomal formulation to New Zealand albino rabbits was greater than 100 mg/kg (brucine content) and there was no skin irritation to integrity and broken skin of New Zealand albino rabbits, while free brucine exhibited acute toxicity to skin and slight irritation to broken skin. It is clear that the toxicity of brucine was reduced after being encapsulated in the liposomes and the prepared LB was a safe formulation, even used on broken skin, which was common in traumatic pains. The in vitro high permeation through the skin and low drug accumulation in skin, which are the inherent advantage of transdermal liposomal delivery system, might contribute to this low dermal toxicity.
The safety evaluation of LB is the prerequisite for the application of brucine, but the question remains as to whether LB keeps the pharmacodynamic actions of brucine after being encapsulated. Previous studies have shown that brucine was mainly responsible for the analgesic and anti-inflammatory effects of the seeds of Strychnos nux-vomica[2]. Our present study reevaluated the analgesic and anti-inflammatory activities using the acetic acid-induced writhing test and xylene-induced mouse ear edema models after brucine was encapsulated in liposomes. The acetic acid-induced writhing test and xylene-induced mouse ear edema are valid and reliable models of nociception and edema. The trends of analgesic and anti-inflammatory activities of LB and free brucine, as shown in Table 3–6, are similar. The two medicaments, whether encapsulated or not, all have analgesic and anti-inflammatory effects. But in contrast, the analgesic and anti-inflammatory efficiency of the LB at dosage ranging from 1.5 mg/kg to 6 mg/kg are obvious better than that of the free brucine. Additionally, LB was exhibited in a dose-dependent manner in the 2 experiments and maintained significantly longer analgesic and anti-inflammatory efficacy. These results illustrated that LB not only keeps the pharmacodynamic effects of brucine, but also pronouncedly increases its bioavailability, which may be attributable to the sustained-release and enhanced-penetration of liposomes. From these findings, LB, prepared in our study, can be confirmed as a transdermal formulation, which exerts a potential topical and pantosomatous therapeutical effect.
In conclusion, the transdermal preparation of LB succeeded in achieving attenuation, synergia and sustained-release of brucine. It represents a novel, safe and effective transdermal therapeutic formulation for relieving acute and chronic pains such as arthritic and traumatic pain. These encouraging results exhibit a good basis for the further development of poisonous traditional Chinese medicine or Western medicine.
References
- Chinese Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China Volume I. Beijing: Chemical Industry Press; 2005.
- Yin W, Wang TS, Yin FZ, Cai BC. Analgesic and anti-inflammatory properties of brucine and brucine N-oxide extracted from seeds of Strychnos nux-vomica. J Ethnopharmacol 2003;88:205-14.
- Zhu YN, Bao MZ, Chen D, Liu H, Li JT, Yan HT, et al. Study on the toxicity of brucine in mice. Henan Med Res 1994;3:107-9.
- Torchilin V, Weissing V,Editors. Liposomes (A Practical Approach). Oxford: IRL Press/Oxford University Press; 1990.
- Srinath P, Vyas SP, Diwan PV. Preparation and pharmacodynamic evaluation of liposomes of indomethacin. Drug Dev Ind Pharm 2000;26:313-21.
- Singh AK, Das J. Liposome encapsulated vitamin A compounds exhibit greater stability and diminished toxicity. Biophys Chem 1998;73:155-62.
- Atif A, Krishna KP, Farhan JA, Yashomati D, Zeenat IK, Divya V. Comparative efficacy of liposome-entrapped amiloride and free amiloride in animal models of seizures and serum potassium in mice. Eur Neuropsychopharmacol 2007;17:227-9.
- Schmid MH, Korting HC. Liposomes: a drug carrier system for topical treatment in dermatology. Crit Rev Ther Drug Carrier Syst 1994;11:97-118.
- Touitou E, Junginger HE, Weiner ND, Nagai N, Mezei M. Liposomes as carriers for topical and transdermal delivery. Int J Pharm 1994;83:1189-203.
- Zhang GF, Zhu JB, Zhuang Y, Hu J. Study on optimize the preparation and formulation of nimodipine-containing nanolipo-some. J Chin Pharm Univ 2003;34:25-8.
- Lboutounne H, Guillaume YC, Michel L, Makki S, Humbert PH, Millet J. Study and development of encapsulated form of 4, 5, 8-trimethylpsoralen for topical drug delivery. Drug Dev Res 2004;61:86-94.
- Isbrucker RA, Edwards JA, Wolz E, Davidovich A, Bausch J. Safety studies on epigallocatechin gallate (EGCG) preparations. Part 2: Dermal, acute and short-term toxicity studies. Food Chem Toxicol 2006;44:636-50.
- Michael S, Taharat Y, Anthony EK, Aimee LL, Jason B, Karrie AB, et al. Safety and toxicological evaluation of a novel niacin-bound chromium (III) complex. J Inorg Biochem 2005;99:2161-83.
- Tang XC, Lin ZG, Cai W, Chen N, Shen L. Anti-inflammatory effect of 3-acetylaconitine. Acta Pharmacol Sin 1984;5:85-9.
- Koster R, Anderson M, De Beer EJ. Acetic acid for analgesic screening. Fed Proc 1959;18:412-6.
- Verma DD, Verma S, Blume G, Fahr A. Particle size of liposomes influences dermal delivery of substances into skin. Int J Pharm 2003;258:141-51.