
Influence of irbesartan on expression of ILK and its relationship with epithelial-mesenchymal transition in mice with unilateral ureteral obstruction
Introduction
Tubulointerstitial fibrosis (TIF) is a common final pathway leading to end-stage renal disease, irrespective of the nature of the initial renal injury. The process of TIF involves the loss of renal tubuli and the accumulation of extracellular matrix (ECM) proteins. Recent studies have highlighted the role of tubular epithelial cells in mediating TIF through epithelial–mesenchymal transition (EMT); the latter is characterized by the loss expression of E-cadherin and the increasing expression of α-smooth muscle actin (α-SMA)[1]. The selective blockade of tubular EMT, due to the preservation of tubular basement membrane integrity in tPA–/– mice, protects the kidney from developing fibrotic lesions after obstructive injury[2]. These observations documented the crucial importance of tubular EMT in the onset and progression of chronic renal fibrosis which eventually results in end-stage renal failure.
Tubular EMT is regulated by numerous growth factors and hormones in different ways. Among them, transforming growth factor-β1 (TGF-β1) is of most importance [3]. Li et al suggested that a linear pathway might exist that couples TGF-β1, Smad signaling, the integrin-linked kinase (ILK), and tubular EMT[4].
The ILK, an intracellular serine/threonine protein kinase that interacts with the cytoplasmic domains of β-integrins and numerous cytoskeleton-associated proteins[5], has been shown to be involved in the regulation of a number of integrin-mediated processes, including cell adhesion, cell phenotypic changes, gene expression, and ECM deposition[6]. Recent studies have implicated ILK dysregulation in the development of several chronic glomerular diseases. For instance, Kretzler et al identified the ILK as a candidate downstream effector of proteinuria in patients with congenital nephritic syndrome[7]. The abundant expression of the ILK is associated with progressive focal segmental glomerulosclerosis[8]. Likewise, analyses of glomerular mesangial cells and human kidney tissues suggest that the ILK is involved in mesangial matrix expansion in response to hyperglycemia in diabetic nephropathy[9]. However, questions remain as to whether the dysregulation of the ILK is also implicated in chronic renal interstitial fibrosis, specifically, how ILK induction leads to the activation of matrix-producing fibroblasts in the fibrotic kidney.
Recent studies have suggested that the activation of the intrarenal renin angiotensin system (RAS) might play an important role in progressive renal fibrosis[10]. Angiotensin II (Ang II) is not only served as a vasoactive agent, but also as a true cytokine with an active role in renal pathology[11]. The activation of the Angiotensin II type 1 (AT1) receptor results in vasoconstriction, stimulation of growth, and the activation of fibroblasts and myocytes[12], thereby largely mediating the fibrogenesis process. Irbesartan, a new type 1 Ang II receptor blocker (ARB), has been proven to be renal protective in both diabetic and non-diabetic nephropathy[13]. Although its renal protective role through the reduction of proteinuria and blood pressure has been noted, more attention has been paid to its mechanism on renal structural remodeling, and an important point of note is the influence of Ang II on renal tubular EMT. The purpose of this study was to investigate the influence of irbesartan on the expression of recently recognized ILK and its relationship with EMT in mice with unilateral ureteral obstruction (UUO).
Materials and methods
Animal model Male CD-1 mice weighing 18–25 g (10 weeks old) were obtained from the Institute of Experimental Animals, Nanjing Jinling Hospital, Nanjing University (Nanjing, China). The mice were randomly divided into 3 groups: control (sham operation, n=20), UUO (n=40), and UUO with irbesartan treatment (UUO+irbesartan, n=40). UUO was performed using an established procedure as described in a previous study[14]. Irbesartan was given at a dose of 50 mg/kg body weight per day by gavage. The experimental animals in the control group received the same volume of vehicle (0.9% saline solution). The mice were killed on d 1, 3, 7, and 14, respectively, and the kidneys were removed for pathological study. The kidneys for the immunohistochemical study were fixed in 10% phosphate buffered formalin, followed by paraffin embedding. The tissues for the protein and mRNA extractions were snap-frozen in liquid nitrogen and stored at -80 °C.
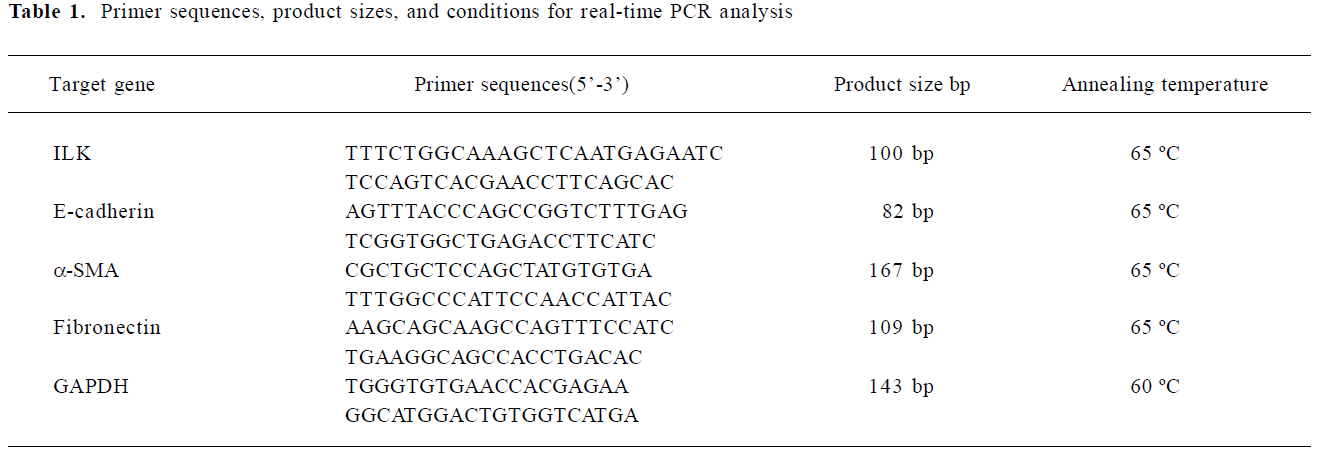
Light microscopic study The paraffin-embedded sections (3 µm) were stained with Masson trichrome. Each of the 30 random, non-overlapping high power fields (magnification ×400) was transferred to the screen of a computer. The percentage fractional area of interstitial fibrosis was determined by taking the number of grid points staining for the blue fibrotic tissue in the interstitium (excluding areas within glomeruli and tubules) using Image Pro Plus software (Media Cybernetics, San Diego, CA, America)
Immunohistochemistry The sections were placed into xylene to remove the paraffin wax and hydrated in graded ethanol. The immunohistochemical detection for the ILK, E-cadherin, and α-SMA was performed according to the streptavidin-biotin immunoperoxidase method SP method (SP kit, Maixin Biotechnology, Fuzhou, China). Briefly, the tissue sections were incubated with pepsin for 5 min to expose the antigen and then endogenous peroxidase activity was blocked for 30 min at room temperature. After washing the sections in phosphate-buffered saline (PBS) 3 times, the sections were then incubated with the primary antibody (rabbit polyclonal anti-ILK [1:100], anti-E-cadherin [1:100], and anti-α-SMA [1:100]) overnight at 4 °C. Each section was washed 3 times in PBS and then incubated with the second antibody for 1 h at room temperature and then reagent D for 30 min. The end compounds reacted with the AEC (3-amino-9-ethy1-carbazole) or DAB (Diaminobenzidin) reagents. The slides were counterstained with hematoxylin and mounted. As a negative control, the primary antibody was replaced with PBS. The positive-stained area was calculated using Image Pro Plus software described earlier and then averaged.
Western blot analysis 50 mg renal tissues from each group were homogenized using an electric tissue grinder in 0.5 mL RIPA buffer [50 mmol/L Tris-HCl (pH 7.5), 1 mmol/L EGTA, 140 mmol/L NaCl, and 1.0% Nonidet P-40], containing 1 µg/mL leupeptin and aprotinin, 1 mmol/L sodium fluoride, 0.1 mmol/L sodium orthovanadate, and 1.0 mmol/L phenyl-methylsulfonyl fluoride. The protein concentrations of all the tissue extracts were determined using the bicinchoninic acid BCA protein assay kit (Pierce, Rockford, IL, America). The samples were heated at 100 °C for approximately 3–4 min before loading and separated on precast 10% SDS–PAGE. After the proteins were electrotransferred to a PVDF (polyvinylidene difluoride) membrane, non-specific binding to the membrane was blocked for 1 h at room temperature with 5% BSA. The membranes were then incubated for 2 h at room temperature with various primary Ab (anti-ILK antibody, Cell Science, USA; dilution of 1:500) in blocking buffer containing 5% milk at the dilutions specified by the manufac-turers. Following extensive washing in TBS buffer, the membranes were incubated with HRP (Peroxidase, Horseradish)-conjugated secondary Ab (1:10000) for 1.5 h at room temperature in 5% non-fat milk dissolved in TBS. The membranes were then washed with TBS buffer and the signals were visualized using the ECL(enhanced chemilumine-scene) system (KangChen KC-420,Shanghai, China) as recommended by the manufacturer. Images were analyzed by Image-J software (NCBI).
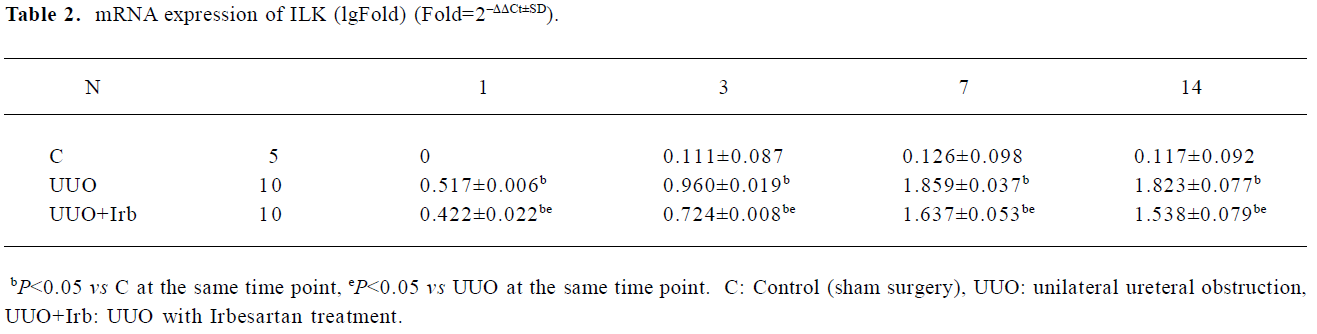
Extraction of total RNA and semiquantitative real-time PCR analysis The total RNA was isolated with the Trizol 1-step method from renal tissues, dissolved in DEPC (diethyl pyrocarbonate) -treated water, quantitated by spectrometry at 260 nm, and stored at -80 °C until assay. One microgram of total RNA from each sample was used for reverse transcription (RT) using the TaKaRa SYBR RT-PCR kit (TaKaRa Biotechnology, Dalian, Jiangsu, China) with 1× M-MLV(Moloney Murine Leukemia Virus) buffer, deoxy-ribonucleoside triphosphate (dNTP) mixture (0.5 mmol/L), oligo dT primer (25 pmol), M-MLV RTase (50 U), RNase inhibitor (10 U), and RNase-free water up to 10 µL at 42 °C for 15 min, 99 °C for 5 min, and then 5 °C for 5 min. For the quantification of mRNA for connective tissue growth factor CTGF, the ILK, E-cadherin, α-SMA, and fibronectin, LightCycler-FastStart DNA Master SYBR Green I system was used (GeneAmp, Foster City, CA, USA). Sequences of the mouse primer pairs are listed in Table 1. The PCR reaction mixture was filled up with SYBR premix Ex Taq (TaKaRa Biotechnology, Dalian, Jiangsu, China) and distilled water to a final volume of 20 uL. PCR reactions were carried out in a real-time PCR cycler (LightCycler) and analyzed using Roche Molecular Biochemicals LightCycler software version 3.5 (Roche Diagnostics, Roche Molecular Biochemicals, Mannheim, Germany). The program was optimized and performed finally as denaturation at 95 °C for 10 min, followed by 40 cycles of amplification (Table 1). The temperature ramp rate was 20 °C/s. At the end of each extension step, the fluorescence of each sample was measured to allow the quantification of the PCR product. After completion of the PCR, the melting curve of the product was measured by temperature gradient from 60 to 95 °C at 0.2 °C/s with continuous fluorescence monitoring to produce a melting profile of the primers. The amount of PCR products was normalized with GAPDH to determine the relative expression ratios for each mRNA in relation to the control group called time zero. Target gene=2–ΔΔCt×Control, ΔΔCt=(Cttarget gene–Ctreference gene)treat group–(Cttarget gene–Ctreference gene)control group RT-PCR experiments were repeated 3 times under identical conditions to verify the results.
Full table
Statistical analysis All the data were shown as mean±SD and analyzed by one-way ANOVA using SPSS software version 11.5 (SPSS, Chicago, IL, USA). P-values <0.05 were accepted as statistically significant.
Results
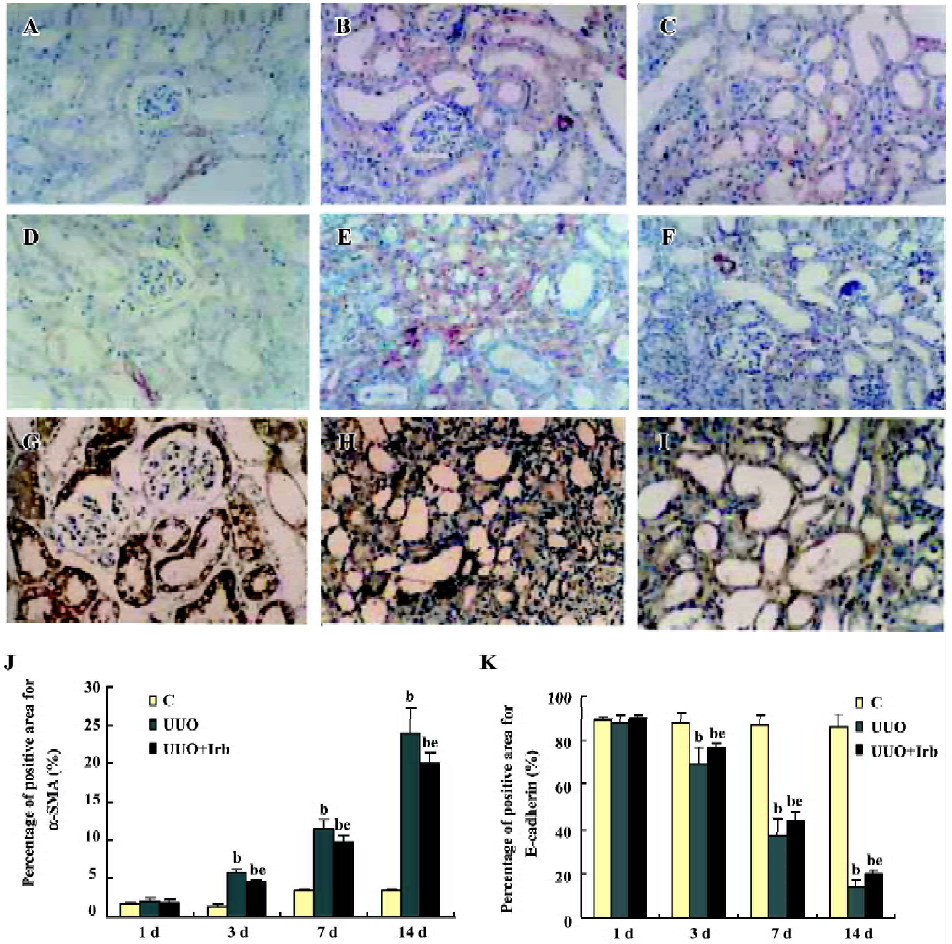
Influence of irbesartan on renal TIF in mice with UUO It was shown by Masson staining that there was no histological abnormality of the kidneys in the sham group. Cellular swelling could be detected in part of the tubular epithelial cells 1 d after the surgery in UUO mice. At d 3, vacuole degeneration, tubular expansion, and leucocyte infiltration could be detected. Tubular atrophy, more infiltration of leucocytes, the expanded tubular interstitial volume, and the over accumulation of the extracellular matrix occurred 7 d after the surgery. Fibrosis could be detected in the renal interstitial area at d 7 and was more severe at d 14 when almost all of the tubules were destroyed. Irbesartan could significantly inhibit renal pathological changes and TIF at different time-points (Figure 1).
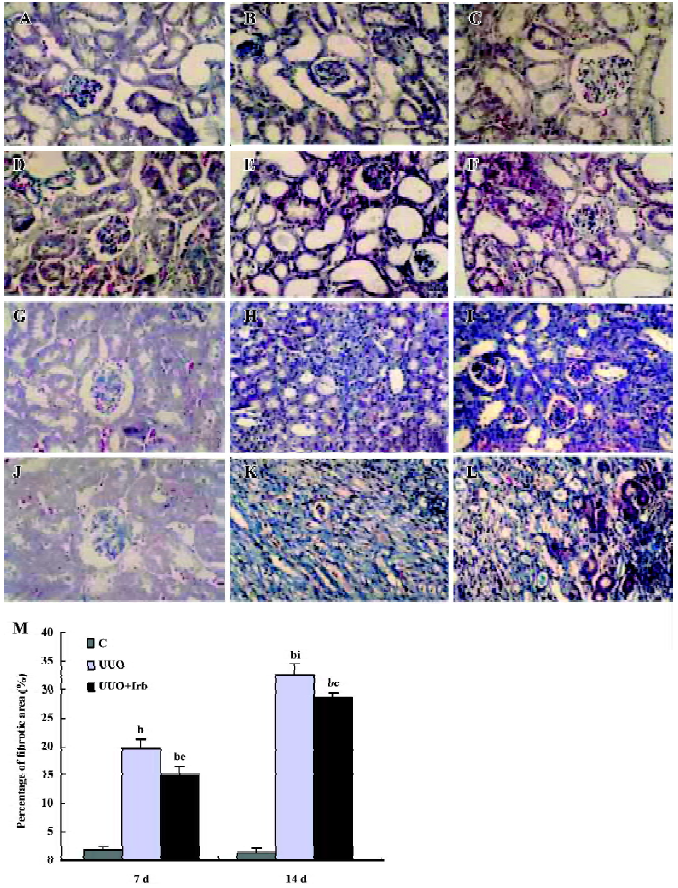
Influence of irbesartan on the expression of the ILK To unravel the mechanism by which irbesartan inhibits tubular EMT, we investigated the effects of blockade of Ang II on the ILK expression. The ILK was mainly expressed in the podocytes of the control group (Figure 2A); a significant increase in the protein and mRNA expressions were detected in mice kidneys with UUO 1 d after the surgery reaching a peak at d 7 (Figures 2B, 2D, 3; Table 2). When severe renal TIF was formed at d 14, both the protein and mRNA expressions of the ILK appeared to decline compared to that at d 7 (Figures 3C, 4; Table 3). Irbesartan could significantly inhibit the expression of the ILK at the protein and mRNA levels (Figures 2C, 2D, 3; Table 2).
Full table
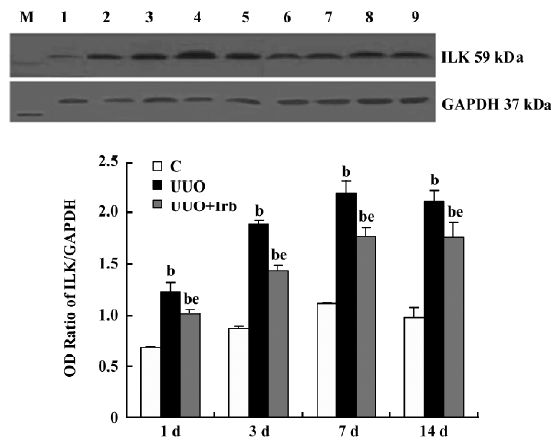
Full table
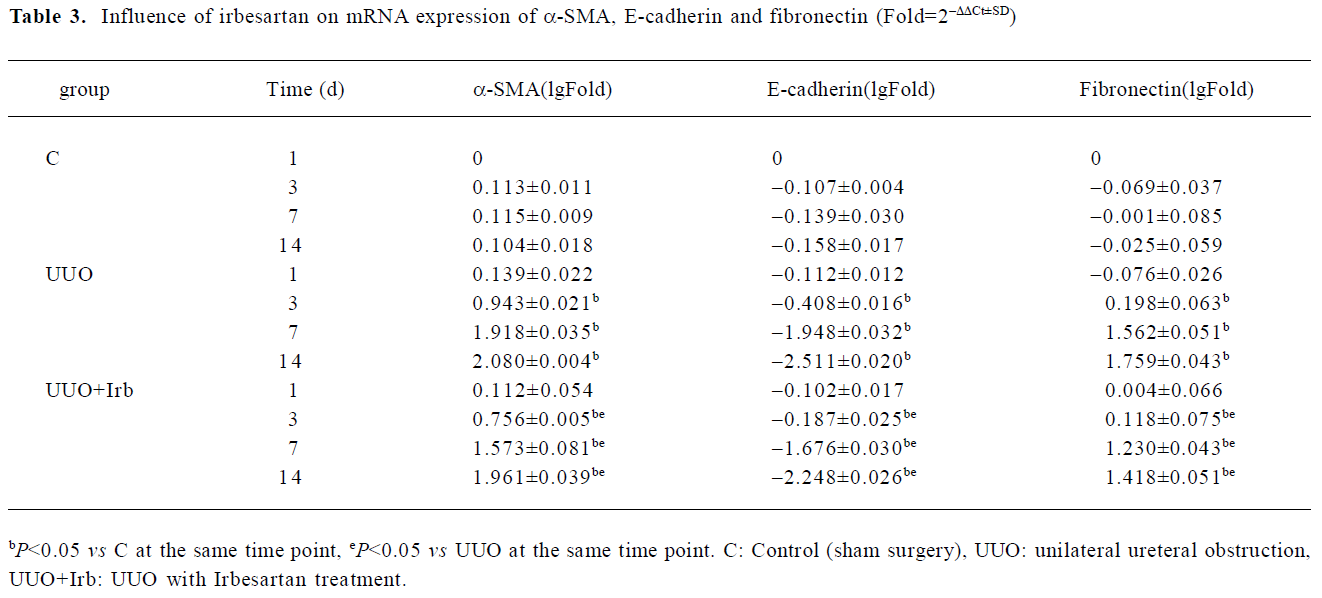
Influence of irbesartan on renal tubular EMT It was shown that a positive reaction to the antibody of α-SMA was detected exclusively in vascular smooth muscle cells in the control group (Figure 4A, 4D). In the mice in the d 3 group, immunostaining demonstrated that de novo α-SMA was expressed in renal tubular epithelial cells (TEC) (Figure 4B). As the disease progressed, the number of α-SMA-positive cells increased, which were mainly expressed in the fibrotic area (Figure 4E). The mRNA expression of α-SMA detected by real-time PCR was continuously upregulated from d 3 onwards (Table 3). As shown in Figure 4G, E-cadherin, an epithelial marker, was expressed in almost all of the epithelial cells within glomeruli and tubuli. Three days after UUO, the expression of E-cadherin significantly decreased, and was continuously downregulated with lesion progression (Figure 4H, 4K; Table 3). Irbesartan could significantly inhibit the expression of α-SMA and the loss of E-cadherin at different time-points. One of the cellular consequences of EMT is the production of massive interstitial matrix components. Along with this, we examined the effects of irbesartan on fibronectin expression in the kidneys. A real-time PCR analysis revealed that the fibronectin mRNA expression significantly increased from d 3 onwards, and irbesartan could significantly abrogate this effect (Table 3).
Correlation between the ILK and EMT markers To clarify the involvement of the ILK in the development of EMT, we examined the correlation between various proteins. Among the parameters tested by immunohistochemistry from d 1 to d 7 after the surgery, the protein expression of the ILK was positively correlated with that of α-SMA (r=0.88, P<0.01), but negatively with E-cadherin (r=-0.87, P<0.01).
Discussion
A number of studies have shown that the activation of the RAS plays an important role in mediating the progression of renal diseases. As the main effector of the RAS, Ang II exerts its vasoconstricting effect predominantly on the post-glomerular arterioles, thereby increasing glomerular hydraulic pressure and promoting glomerular ultrafiltration, which may contribute to the onset and progression of chronic renal damage[13]. Furthermore, several lines of evidence have demonstrated that Ang II might also accelerate renal damage by its non-hemodynamic effects[14], such as increasing the production of reactive oxygen species, the upregulation of cytokines, cell adhesion molecules, and profibrotic growth factors leading to renal fibrosis. Ang II acts through its binding to 2 specific receptors, AT1 and AT2[12]. AT1 is responsible for most of the pathophysiological actions of Ang II. The ARB are highly effective and well-tolerated antihypertensive medications, and recent clinical trials have shown that these agents have renoprotective effects beyond lowering blood pressure. Irbesartan is a new type of ARB. Two important studies that included more than 1500 patients with type 2 diabetes and overt nephropathy demonstrated that irbesartan reduced the relative risk of micro-albuminuria progression to macroproteinuria compared to amlodipine, but its exact mechanism for renoprotection is still uncertain.
Fibroblasts are the principal effectors in the development of TIF. Under pathological conditions, they can rapidly proliferate, produce interstitial collagens, and contribute to the progression of tissue fibrosis[1]. Interstitial fibroblasts are much more heterogeneous than expected and they might come from interstitial resident fibroblasts, circulating fibrocytes, EMT-derived fibroblasts, and bone marrow-derived fibroblasts[15]. Among them, the most plausible explanation for the replenishment of fibroblasts during fibrogenesis is EMT. The strongest support for a pathogenetic role of EMT is derived from in vivo observations using bone marrow chimeras and transgenic reporter mice in which fibroblasts were specifically labeled with green fluorescent protein. Taking this approach, Iwano et al[16] showed that during renal fibrogenesis, interstitial kidney fibroblasts are derived in small numbers from bone marrow and in large numbers from local EMT. Such an accumulation of EMT-derived fibroblasts associated with tubular decondensation and atrophy is considered to be a key determinant of renal fibrosis during chronic injury.
The feature of EMT is cell loss of epithelial adhesion properties and de novo expression of α-SMA and actin reorganization; disruption of tubular basement membrane; and enhanced cell migration and invasion[17]. In this study, we found that the expression of E-cadherin for both mRNA and protein significantly decreased from d 3 onwards after the surgery in the kidneys of mice with UUO. Under normal conditions, tubular epithelial cells are tightly connected to each other to form an integrated epithelial sheet through various cell adhesion mechanisms. E-cadherin, the well-characterized adhesion receptor found within adherens type junctions, plays an essential role in maintaining the structural integrity of renal epithelia and its polarization. The suppression of E-cadherin expression is regarded as a key step that precedes other major events during tubular EMT and will presumably lead to the destabilization of epithelial sheet integrity, making cells ready to lose polarity, to dissociate from their neighbors, and migrate. α-SMA, the hallmark of myofibroblasts, may provide a structural foundation not only for defining the morphology of the transformed cells, but also for them to migrate, invade, and even acquire the capacity for contractility. α-SMA-positive TEC are of a stable, terminally-differentiated, irreversible, transformed cell type[18]. Positive staining for α-SMA was detected exclusively in vascular smooth muscle cells in the control group. At d 3 after the surgery, de novo α-SMA protein expression could be detected in renal TEC and the mRNA expression of α-SMA, detected by real-time PCR, was continuously upregulated from d 3 onwards. The de novo expression of α-SMA and the loss of E-cadherin indicate that the mesenchymal program gene is turned on when the epithelial program gene has been switched off.
Another hallmark for tubular EMT is that the tubular epithelial cells begins to overproduce ECM components and properly assembles in the extracellular compartment, leading to excessive accumulation of ECM, causing massive tissue fibrosis as seen in diseased kidneys. The present study showed that the mRNA expression of fibronectin significantly increased from d 3 onwards. Irbesartan significantly inhibited the expression of α-SMA and fibronectin and reversed the loss of E-cadherin, suggesting that irbesartan could reverse tubular EMT. EMT is regulated by many factors, such as TGF-β[19], Ang II, the epidermal growth factor, fibroblast growth factor-2[20], and reactive oxygen species[21,22], among which TGF-β is believed to be of most importance. Liu et al demonstrated that Ang II acts as a strong promoter that dramatically potentiates the ability of TGF-β1 to induce EMT in tubular epithelial cells[23]. We recently demonstrated that Ang II potentiated tubular cells developing EMT via the production of CTGF[24]. In the present study, we also demonstrated that the AT1 receptor antagonist irbesartan significantly inhibited tubular EMT, but the mechanism of this process is currently under investigation. The ILK is an intracellular serine/threonine protein kinase that interacts with the cytoplasmic domains of β-integrins and numerous other cytoskeleton-associated proteins. Differential regulation of the ILK has been observed during the pathogenesis of glomerular disease and TIF. In outside–in signaling, ILK mediates the response of renal cells to alterations in the matrix and growth factor environments. In inside–out signaling, ILK transduces inflammatory and oxidative stress responses into decreased matrix attachments[25]. The downstream signaling of ILK activates the Wnt pathway with a switch towards a proliferative, mesenchymal phenotype[26]. In this process, ILK influences the actin cytoskeleton, resulting in shape change and focal adhesion dysfunction observed in podocyte failure and TIF. Recent studies found that TGF-β could induce the Smad-dependent expression of ILK in tubular epithelial cells. ILK plays a critical role in TGF-β-induced EMT[8]. Hepatocyte growth factor, a well-known antifibrotic factor, was also shown to have an antifibrotic effect through the inhibition of ILK and the blockade of EMT. However, the relationship between Ang II and ILK in the process of EMT has not been addressed previously. In this study, we first demonstrated that irbesartan could significantly inhibit the expression of ILK both at the mRNA and protein levels, and the protein expression of ILK is positively correlated with that of α-SMA, but negatively correlated with that of E-cadherin. It is therefore suggested that there might be an independent pathway of Ang II-ILK in mediating EMT.
Taken together, our findings shed new light on the mechanism underlying Ang II in mediating TIF. Irbesartan exerted a renal protective effect in mice with UUO through inhibiting renal tubular EMT by downregulating the expression of ILK. These data provide further evidence for preventing the development of TIF by blocking the intrarenal RAS.
References
- Faulkner JL, Szcykalski LM, Springer F, Barnes JL. Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am J Pathol 2005;167:1193-205.
- Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C, et al. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest 2002;110:1525-38.
- Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, et al. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 1999;56:1455-67.
- Li Y, Yang J, Dai C, Wu C, Liu Y. Role of integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest 2003;112:503-16.
- Wu CY. The PINCH-ILK-parvin complexes: assembly, functions and regulation. Biochim Biophys Acta 2004; 1692: 55–62.
- Ruiz-Torres MP, Lopez-Ongil S, Griera M, Diez-Marques ML, Rodriguez-Puyol M, Rodriguez-Puyol D. The accumulation of extracellular matrix in the kidney: consequences on cellular function. J Nephrol 2005;18:334-40.
- Kretzler M, Teixeira VP, Unschuld PG, Cohen CD, Wanke R, Edenhofer I, et al. Integrin-linked kinase as a candidate downstream effector in proteinuria. FASEB J 2001;15:1843-5.
- Guo L, Sanders PW, Woods A, Wu C. The distribution and regulation of integrin-linked kinase in normal and diabetic kidneys. Am J Pathol 2001;159:1735-42.
- Ruiz-Ortega M, Ruperez M, Esteban V, Egido J. Molecular mechanisms of angiotensin II-induced vascular injury. Curr Hypertens Rep 2003;5:73-9.
- Misseri R, Rink RC, Meldrum DR, Meldrum KK. Inflammatory mediators and growth factors in obstructive renal injury. J Surg Res 2004;119:149-59.
- Ruiz-Ortega M, Ruperez M, Esteban V, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney disease. Nephrol Dial Transplant 2006;21:16-20.
- Remuzzi G, Perico N, Macia M, Ruggenenti P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int 2005;68 Suppl 99:S57-65.
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol 2002;283:F861-75.
- Aros C, Remuzzi G. The renin-angiotensin system in progression, remission and regression of chronic nephropathies. J Hyperten 2002;20 Suppl 3:S45-43.
- Zeisberg M, Kaluri R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med 2004;82:175-81.
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341-50.
- Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol 2001;159:1465-75.
- Lan HY. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens 2003;12:25-9.
- Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal trans-formation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs 2005;179:11-23.
- Liu YH. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 2004;15:1-12.
- Bernardo RI, Richard JJ, Jaime HA. Tubulointerstitial damage and progression of renal failure. Kidney Int 2005;68 Suppl 99:S82-6.
- Ha H, Lee HB. Reactive oxygen species and matrix remodeling in diabetic kidney. J Am Soc Nephrol 2003;14:S246-9.
- Yang J, Dai C, Liu Y. Hepatocyte growth factor gene therapy and angiotensin II blockade synergistically attenuate renal interstitial fibrosis in mice. J Am Soc Nephrol 2002;13:2464-77.
- Chen L, Liu BC, Zhang XL, Zhang JD, Liu H, Li MX. Influence of connective tissue growth factor antisense oligonucleotide on angiotensin II-induced epithelial mesenchymal transition in HK2 cells. Acta Pharmacol Sin 2006;27:1029-36.
- Green K, Watt F. Cell-to-cell contact and extracellular matrix. Curr Opin Cell Biol 2004;16:465-9.
- Oloumi A, McPhee T, Dedhar S. Regulation of E-cadherin expression and β-catenin/Tcf transcriptional activity by the integrin-linked kinase. Biochim Biophys Acta 2004; 1691: 1–15.