
N-Acetylcysteine attenuates lipopolysaccharide-induced apoptotic liver damage in D-galactosamine-sensitized mice1
Introduction
Bacterial lipopolysaccharide (LPS) is a toxic component of cell walls of Gram-negative bacteria and is widely present in the digestive tracts of humans and animals[1]. Humans are constantly exposed to low levels of LPS through infection. Gastrointestinal distress and alcohol consumption often increase the permeability of LPS from the gastrointestinal tract into the blood[2]. In humans, nanograms of LPS injected into the blood stream can result in the physiological manifestations of septic shock[3]. The liver is a highly responsive organ in systemic inflammation caused by LPS. Hepatic dysfunction after sepsis is a frequent event that is characterized by the loss of synthetic function and hepatocellular necrosis[4].
D-Galactosamine (GalN) is an amino sugar selectively metabolized by the hepatocyte, which induces a depletion of the uridine triphosphate (UTP) pool and thereby an inhibition of RNA synthesis[5]. When given together with a low sublethal dose of LPS, GalN highly sensitizes animals to develop lethal liver injury mimicking fulminant hepatitis[6]. GalN/LPS-induced liver injury is characterized by apoptosis of hepatocytes, widespread destruction of liver architecture, and erythrocyte agglutination. In the GalN/LPS model, TNF-α is the major mediator leading to apoptotic liver injury[7]. Nitric oxide may also play a role in GalN/LPS-induced apo-ptotic liver injury[8,9]. Recent studies showed that hydrogen peroxide and reduced glutathione depletion sensitize primary mouse hepatocytes to TNF-α-induced apoptosis[10–12]. Furthermore, rosmarinic acid, an exogenous antioxidant, protects against D-GalN/LPS-induced hepatic apoptosis[13], suggesting that reactive oxygen species (ROS) may be involved in TNF-α-mediated apoptotic liver injury.
N-acetylcysteine (NAC) is a glutathione (GSH) precursor and direct antioxidant. As a potent antioxidant, NAC directly scavenges hydrogen peroxide, hydroxyl free radicals, and hypochloric acid in vitro[14]. NAC also decreases free radical levels by increasing GSH synthesis[15,16]. Several studies have indicated that NAC inhibits LPS-induced inducible nitric oxide synthase, TNF-α expression, and NF-κB activity[17,18]. Our earlier studies showed that pretreatment with NAC prevents the LPS-induced downregulation of the pregnane X receptor and P450 3A11 expression in mouse liver, placenta, and fetal liver[19–21]. Furthermore, pretreatment with NAC protects mice against LPS-induced intrauterine fetal death and intrauterine growth retardation[22]. Recently, we found that pretreatment with NAC attenuates acute ethanol-induced liver damage in mice[23]. Clinically, NAC has been successfully used in adult respiratory distress syndrome[24]. In the present study, we investigated the effect of NAC on LPS-induced apoptotic liver damage in GalN-sensitized mice. Our results showed that NAC protects mice against GalN/LPS-induced apoptotic liver injury via its antioxidant and anti-apoptotic effects.
Materials and methods
Chemicals LPS (Escheria coli LPS, serotype 0127:B8), GalN, DL-buthionine-(SR)-sulfoximine (BSO), NAC, and other reagents were purchased from Sigma Chemical Co (St Louis, MO, USA) unless otherwise stated.
Animals and treatments Female CD-1 mice (6–8 week-old, 24–26 g) were purchased from Beijing Vital River Laboratory Animal Co Ltd (Beijing, China) whose foundation colonies were all introduced from Charles River Laboratories (Wilmington, USA, MA) The animals were allowed free access to food and water at all times and were maintained on a 12 h light/dark cycle in a controlled temperature (20–25 °C) and humidified (50%±5%) environment for a period of 1 week before the experiments. All animal procedures followed the guidelines for humane treatment set by the Association of Laboratory Animal Sciences and the Center for Laboratory Animal Sciences at Anhui Medical University (Hefei, China).
To investigate the protective effects of NAC, the mice were administered with NAC (150 mg/kg, ip) 30 min before GalN/LPS (700 mg/10 µg/kg, ip). The doses of NAC used in the present study referred to those used in previous studies[25]. The control mice received saline. Some mice were killed 1.5 h after GalN/LPS administration. Serum was collected for the measurement of TNF-α. The remaining mice were killed 8 h after GalN/LPS. Serum was collected for the measurement of alanine aminotransferase (ALT) and nitrate plus nitrite. The livers were dissected for the measurement of GSH content, caspase-3 activity, DNA extraction, and histological examination.
Evaluation of hepatotoxicity Serum ALT activity was assayed as a marker of hepatotoxicity using a commercially available kit (Nanjing Jiancheng Institute of Biological Engineering and Technology, Nanjing, China) according to the manufacturer’s instructions. A portion of liver was fixed in 10% formalin, processed by standard histological techni-ques, attained with hematoxylin-eosin (HE), and examined for morphological evaluation of liver injury.
DNA fragmentation analysis The liver tissues were homogenized and incubated in 100 mmol/L Tris-HCl (pH 8.0), 25 mmol/L EDTA, 0.5% SDS, and 0.1 µg/mL proteinase K at 60 °C for 3 h. DNA was extracted with phenol/chloroform (1:1) and chloroform/isoamyl alcohol (1:24). The extracted DNA was precipitated and digested in 10 mmol/L Tris-HCl (pH 5.0) containing 1 mmol/L EDTA and 10 µg RNase for 1 h at 37 °C. 5 µg DNA per sample was electrophoretically separated on 1.5% agarose gel containing 0.5 µg/mL ethidium bromide. The DNA pattern was examined by UV transillumination.
Determination of caspase-3 activity The hepatic caspase-3 activity was determined based on a colorimetric method[26]. Briefly, the liver homogenates were prepared in lysis buffer containing 100 mmol/L HEPES (N-2-hydroxyethylpiperazine-N-2-ethane-sulfonic acid), pH7.5, 20% (v/v) glycerol, 0.5 mmol/L EDTA, and 5 mmol/L dithiothreitol. The protease assay mixture included 240 µL reaction buffer (100 mmol/L HEPES, pH 7.5, 20% glycerol, 0.5 mmol/L EDTA, and 5 mmol/L dithio-threitol), 30 µL of 1 mmol/L acetyl-Asp-Glu-Val-Asp-p-nitroanilide in DMSO, and 30 µL liver homogenates. The samples were incubated at 37 °C for 1 h, and the enzyme-catalyzed release of p-nitroanilide was measured at 405 nm in an ultra-microplate reader (Bio-Tek instruments, Winnoski, Vermont, USA). Caspase-3 activity was expressed as nmol/mg protein. The protein content was measured according to the method of Lowry et al[27].
Determination of GSH content GSH was determined based on the method of Griffith[28]. Proteins of 0.4 mL liver homogenates were precipitated by the addition of 0.4 mL of a metaphosphoric acid solution. After 40 min, the protein precipitate was separated from the remaining solution by centrifugation at 4200×g at 4 °C for 5 min. 400 µL of the supernatant was combined with 0.4 mL of 300 mmol/L Na2HPO4, and the absorbance at 412 nm was read against a blank consisting of 0.4 mL supernatant plus 0.4 mL H2O. Then, 100 µL DTNB (5,5-dithio-bis-2-nitrobenzoic acid) (0.02% [w/v], 20 mg DTNB in 100 mL of 1% sodium citrate) was added to the blank and sample. The absorbance of the sample was read against the blank at 412 nm. The GSH content was determined using a calibration curve prepared with an authentic sample. The GSH values were expressed as nmol/mg protein. The protein content was measured according to the method of Lowry et al[27].
Analysis of serum nitrite plus nitrate concentration The stable end products of L-arginine-dependent nitric oxide synthesis and nitrate plus nitrite, were measured in the serum using a colorimetric method based on the Griess reaction[29]. Briefly, aliquots of serum were added to 35% sulfosalicylic acid and vortexed every 5 min for 30 min to depro-teinize the samples. The samples were then centrifuged at 10 000×g at 4 °C for 15 min. An aliquot of the supernatant was taken for the nitrite plus nitrate analysis. Twenty microliters of the serum sample was mixed with 20 µL of 0.31 mol/L phosphate buffer, pH7.5, 10 µL of 0.1 mmol/L FAD (flavin adenine dinucleotide), 10 µL of 1 mmol/L NADPH, 10 mL of nitrate reductase (10 U/mL), and 30 µL of ddH2O in a 96-well plate. The reaction was allowed to proceed for 1 h in dark. To each sample, 1 µL of lactate dehydrogenase (1500 U/mL) and 10 µL of 100 mmol/L pyruvic acid were added and incubated for 15 min at 37 °C. The samples were then mixed with an equivalent volume of Griess reagent and incubated for an additional 10 min at room temperature. Nitrite levels were determined colorimetrically at 550 nm with a Universal microplate reader (Bio-Tek Instruments, Winnoski, Vermont, USA) and a sodium nitrite standard curve.
Measurement for TNF-α Serum TNF-α levels were measured by ELISA (R&D, Minneapolis, MN, USA), following the manufacturer’s instructions.
Statistical analysis Quantified data were expressed as mean±SEM. ANOVA and the Student-Newman-Keuls post-hoc test were used to determine differences between the treated animals and the control and statistical significance.
Results
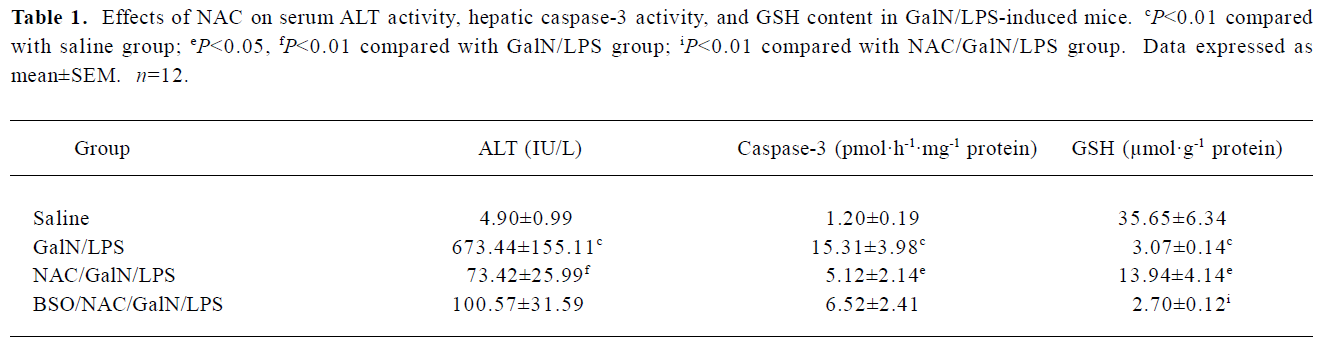
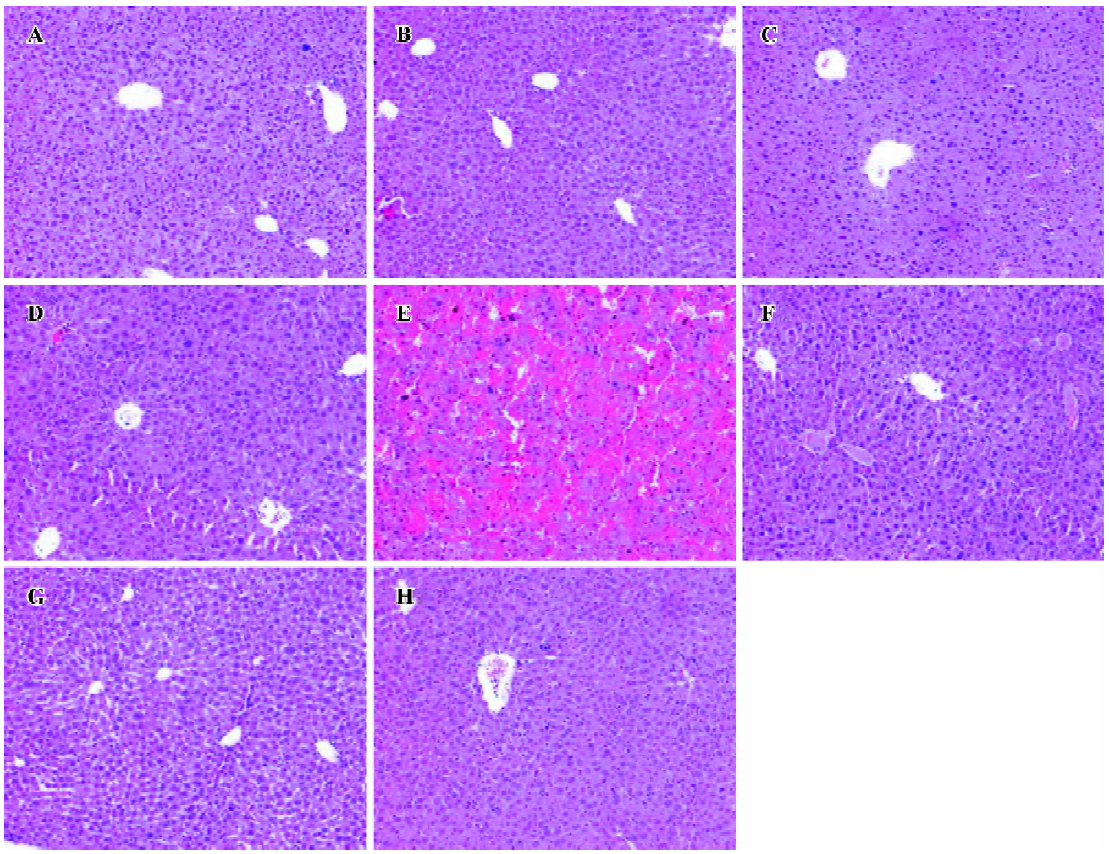
Effects of NAC on GalN/LPS-induced liver injury To investigate the effects of NAC pretreatment on GalN/LPS-induced apoptotic liver injury, the mice were pretreated with NAC before GalN/LPS. The results showed that neither GalN nor LPS alone is lethal at low concentrations. Co-injection of GalN and LPS into mice resulted in a lethal liver injury mimicking fulminant hepatitis, in which 6 of the 10 mice died within 12 h. Serum aspartate ALT activity was markedly increased at 8 h after GalN/LPS (Table 1). A histological examination showed massive necrosis of parenchymal hepatocytes with marked hemorrhage in the liver of GalN/LPS-treated mice (Figure 1E). Pretreatment with NAC significantly attenuated GalN/LPS-induced elevation of serum ALT activities. In addition, GalN/LPS-induced histological injury was obviously improved in NAC-pretreated mice (Figure 1F). To investigate the role of GSH on NAC-mediated protection against GalN/LPS-induced liver injury, the mice were pretreated with BSO, an inhibitor of GSH synthesis, to inhibit hepatic GSH synthesis. The results showed that the protective effects of NAC pretreatment on GalN/LPS-induced liver injury were not counteracted by BSO (Table 1; Figure 1H).
Full table
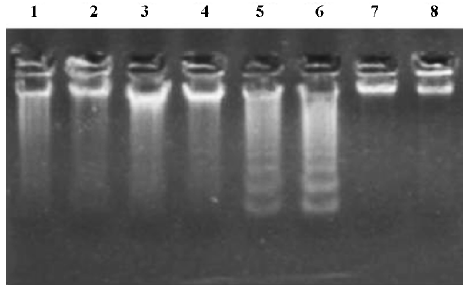
Effects of NAC on GalN/LPS-induced hepatic apoptosis Hepatic apoptosis was clearly confirmed in the GalN/LPS-treated mice, as indicated by DNA laddering on agarose gel electrophoresis (Figure 2, lanes 5 and 6). NAC pretreatment significantly attenuated GalN/LPS-induced hepatic DNA fragmentation (Figure 2, lanes 3 and 4). The effects of NAC pretreatment on hepatic caspase-3 activities in the GalN/LPS-treated mice were further analyzed. Hepatic caspase-3 activity was markedly increased in the GalN/LPS-treated mice (Table 1). Consistent with the attenuation of DNA laddering, NAC pretreatment significantly inhibited hepatic caspase-3 activities in the GalN/LPS-treated mice. To investigate the role of GSH on NAC-mediated protection against GalN/LPS-induced hepatic apoptosis, the mice were pretreated with BSO, an inhibitor of GSH synthesis, to inhibit hepatic GSH synthesis. The results showed that the inhibitive effects of NAC pretreatment on hepatic caspase-3 activity were not counteracted by BSO. In addition, BSO did not influence the protective effects of NAC pretreatment on GalN/LPS-induced hepatic DNA fragmentation (Figure 2, lanes 1 and 2).
Effects of NAC on LPS-induced GSH depletion The effects of NAC on LPS-induced hepatic GSH depletion are presented in Table 1. The results showed that GalN/LPS dramatically decreased hepatic GSH content. Pretreatment with NAC significantly attenuated hepatic GSH depletion. By contrast, BSO aggravated GalN/LPS-induced hepatic GSH depletion.
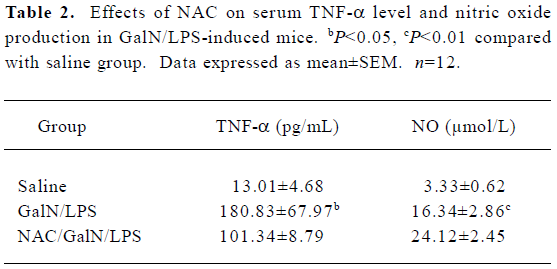
Effects of NAC on serum TNF-α in GalN/LPS-treated mice In the GalN/LPS model, TNF-α is the major mediator leading to liver injury[7]. To investigate the effects of NAC pretreatment on TNF-α production, the mice were pretreated with NAC 0.5 h before GalN/LPS. A preliminary study showed that TNF-α was at peak value at 1.5 h and returned to basal level 6 h after GalN/LPS treatment (data not shown). Therefore, serum TNF-α concentration was measured 1.5 h after GalN/LPS administration. As expected, there was a significant increase in serum TNF-α level in the GalN/LPS-treated mice (Table 2). However, pretreatment with NAC did not decrease the elevation of serum TNF-α level in the GalN/LPS-treated mice.
Full table
Effects of NAC on LPS-induced NO production To investigate the effect of NAC on LPS-induced nitric oxide production in the GalN-sensitized mice, the stable end products of L-arginine-dependent nitric oxide synthesis and nitrate plus nitrite, were measured at 8 h after GalN/LPS. There was a significant increase for serum nitrate plus nitrite levels in the GalN/LPS-treated mice (Table 2). However, pretreatment with NAC had no effect on LPS-evoked elevation of nitric oxide levels.
Discussion
GalN depletes UTP primarily in the liver, resulting in decreased RNA synthesis in hepatocytes[5]. When given together with a low sublethal dose of LPS, GalN highly sensitizes animals to develop lethal liver injury mimicking fulminant hepatitis[6]. In the present study, we showed that co-injection of GalN and LPS into mice produced fulminant hepatitis with severe hepatic congestion, resulting in rapid death. Serum ALT levels were markedly increased 8 h after GalN/LPS, and massive necrosis of parenchymal hepatocytes with marked hemorrhage was observed in the histological sections of the liver from the GalN/LPS-treated mice. NAC is an antioxidant and a GSH precursor. A previous study showed that pretreatment with NAC attenuated organ dysfunction during endotoxemia and protected against LPS-induced liver injury[30]. In the present study, we investigated the effects of pretreatment with NAC on LPS-induced liver injury in GalN-sensitized mice. We found that pretreatment with NAC significantly reduced serum ALT levels in GalN/LPS-treated mice. In parallel, NAC pretreatment significantly attenuated GalN/LPS-induced hepatic necrosis and conges-tion.
GalN/LPS-induced liver injury is characterized by apoptosis of hepatocytes. A previous study showed that treatment with YVAD-CMK (Acetyl-Tyr-Val-Ala-Asp-chloromethy- lketone), a potent tetrapeptide inhibitor of the interleukin (IL)-1β conterting enzyme family, protects from LPS-induced apoptotic liver injury in GalN-sensitized mice[31]. In the present study, hepatic apoptosis was clearly confirmed in the GalN/LPS-treated mice, as indicated by strong DNA laddering on agarose gel electrophoresis. Moreover, co-injection of GalN and LPS significantly increased caspase-3 activity in the mouse liver. NAC is an anti-apoptotic mediator[32,33]. The present study showed that NAC pretreatment significantly inhibited hepatic caspase-3 activity in GalN/LPS-treated mice. NAC pretreatment significantly attenuated GalN/LPS-induced hepatic DNA fragmentation. These results indicate that the protective effect of NAC pretreatment against GalN/LPS-induced apoptotic liver injury might be, at least in part, mediated by its anti-apoptotic effects.
In the GalN/LPS model, TNF-α is the major mediator of liver injury[7]. Several studies have demonstrated that NAC significantly inhibits the LPS-induced release of TNF-α in Kupffer cells[15,34–36]. In the present study, we investigated the effect of NAC pretreatment on GalN/LPS-induced TNF-α production. As expected, LPS significantly increased serum TNF-α level in GalN-sensitized mice. However, the present study showed that the LPS-induced elevation of the TNF-α level was not significantly reduced by NAC pretreatment. Therefore, the present study does not determine whether NAC-mediated protection against GalN/LPS-induced liver injury is due to the inhibition of TNF-α production.
A recent study indicated that ROS are involved in GalN-induced sensitization against TNF-α-induced hepatocyte apoptosis[37]. Concomitant TNF-α exposure and ROS, either extrinsically generated by non-parenchymal or inflammatory cells or intrinsically generated in hepatocytes, may act in concert to promote apoptosis and liver injury[10]. Moreover, rosmarinic acid, an exogenous antioxidant, protects against D-GalN/LPS-induced hepatic apoptosis[13]. In addition, reduced GSH depletion has also been demonstrated to sensitize primary mouse hepatocytes to TNF-α-induced apoptosis[11,12]. NAC is not only a direct antioxidant, but also a GSH precursor. NAC acts as a free radical scavenger, directly scavenges hydrogen peroxide, hypochloric acid, and hydroxyl radical. On the other hand, NAC acts as a precursor of GSH to facilitate intracellular GSH synthesis[14]. In the present study, we analyzed the effects of NAC on hepatic GSH content in GalN/LPS-treated mice. As expected, pretreatment with NAC significantly attenuated GalN/LPS-induced hepatic GSH depletion. However, BSO, an inhibitor of GSH synthesis, did not influence NAC-mediated protection against GalN/LPS-induced apoptotic liver injury, although GalN/LPS-induced hepatic GSH depletion was aggravated by BSO in NAC-pretreated mice. These results suggest that the NAC-mediated protection against GalN/LPS-induced apoptotic liver injury is not attributed to increased GSH synthesis, but most likely due to its strong ROS-scavenging effect.
Nitric oxide plays an important role in GalN/LPS-induced apoptotic liver injury[8,9]. Several studies demonstrated that NAC inhibits LPS-evoked inducible nitric oxide synthase expression and nitric oxide release in macrophages[17,38]. In the present study, we investigated the effects of NAC pretreatment on nitric oxide production. We found that serum nitrate plus nitrite, the stable end products of L-arginine-dependent nitric oxide synthesis, significantly increased at 8 h after GalN/LPS administration. However, NAC pretreatment had no effect on GalN/LPS-evoked nitric oxide production. These results suggest that the protective effect of NAC pretreatment against GalN/LPS-induced apoptotic liver injury is not be mediated by the inhibition of nitric oxide release.
In summary, the present study indicated that pretreatment with NAC has a beneficial effect on GalN/LPS-induced apoptotic liver injury. The protective effect of NAC pretreatment may be mediated by its strong ROS-scavenging and anti-apoptotic effects.
References
- Jacob AL, Goldberg PK, Bloom N, Degenshein GA, Kozinn PJ. Endotoxin and bacteria in portal blood. Gastroenterology 1977;72:1268-70.
- Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol 1991;12:162-9.
- Michie HR, Manogue KR, Spriggs DR, Revhaug A, Dwyer SI, Dinarello A, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med 1988;318:1481-6.
- Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol 2002;283:256-65.
- Decker K, Keppler D. Galactosamine hepatitis: key role of the nucleotide deficiency period in the pathogenesis of cell injury and cell death. Rev Physiol Biochem Pharmacol 1974;71:77-105.
- Galanos C, Freudenberg MA, Reutter W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc Natl Acad Sci USA 1979;76:5939-43.
- Tiegs G, Wolter M, Wendel A. Tumor necrosis factor is a terminal mediator in galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol 1989;38:627-31.
- Liu J, Saavedra JE, Liu T, Song JG, Waalkes MP, Keefer LK. O. (2)-Vinyl 1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate protection against D-galactosamine/endotoxin-induced hepatotoxicity in mice: genomic analysis using microarrays. J Pharmacol Exp Ther 2002;300:18-25.
- Morikawa A, Kato Y, Sugiyama T, Koide N, Chakravortty D, Yoshida T, et al. Role of nitric oxide in lipopolysaccharide-induced hepatic injury in D-galactosamine-sensitized mice as an experimental endotoxic shock model. Infect Immun 1999;67:1018-24.
- Han D, Hanawa N, Saberi B, Kaplowitz N. Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic Biol Med 2006;41:627-39.
- Matsumaru K, Ji C, Kaplowitz N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology 2003;37:1425-34.
- Nagai H, Matsumaru K, Feng G, Kaplowitz N. Reduced glutathione depletion causes necrosis and sensitization to tumor necrosis factor-alpha-induced apoptosis in cultured mouse hepatocytes. Hepatology 2002;36:55-64.
- Osakabe N, Yasuda A, Natsume M, Sanbongi C, Kato Y, Osawa T, et al. Rosmarinic acid, a major polyphenolic component of Perilla frutescens, reduces lipopolysaccharide (LPS)-induced liver injury in D-galactosamine (D-GalN)-sensitized mice. Free Radic Biol Med 2002;33:798-806.
- Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med 1989;6:593-7.
- Neuschwander-Tetri BA, Bellezzo JM, Britton RS, Bacon BR, Fox ES. Thiol regulation of endotoxin-induced release of tumour necrosis factor alpha from isolated rat Kupffer cells. Biochem J 1996;320:1005-10.
- Song M, Kellum JA, Kaldas H, Fink MP. Evidence that glutathione depletion is a mechanism responsible for the anti-inflammatory effects of ethyl pyruvate in cultured lipopolysaccharide-stimulated RAW 264.7 cells. J Pharmacol Exp Ther 2004;308:307-16.
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. N-acetyl cysteine inhibits induction of no production by endotoxin or cytokine stimulated rat peritoneal macrophages, C6 glial cells and astrocytes. Free Radic Biol Med 1998;24:39-48.
- Verhasselt V, Vanden-Berghe W, Vanderheyde N, Willems F, Haegeman G, Goldman M. N-acetyl-L-cysteine inhibits primary human T cell responses at the dendritic cell level: association with NF-kappaB inhibition. J Immunol 1999;162:2569-74.
- Chen YH, Wang JP, Wang H, Sun MF, Wei LZ, Wei W, et al. Lipopolysaccharide treatment downregulates the expression of the pregnane X receptor, cyp3a11 and mdr1a genes in mouse placenta. Toxicology 2005;211:242-52.
- Xu DX, Wei W, Sun MF, Wu CY, Wang JP, Wei LZ, et al. Kupffer cells and ROS partially mediate LPS-induced down-regulation of nuclear receptor Pregnane X receptor and its target gene CYP3A in mouse liver. Free Radic Biol Med 2004;37:10-22.
- Xu DX, Chen YH, Wang JP, Sun MF, Wang H, Wei LZ, et al. Perinatal lipopolysaccharide exposure downregulates pregnane X receptor and cyp3a11 expression in fetal mouse liver. Toxicol Sci 2005;87:38-45.
- Xu DX, Chen YH, Wang H, Zhao L, Wang JP, Wei W. Effect of N-acetylcysteine on lipopolysaccharide-induced intra-uterine fetal death and intra-uterine growth retardation in mice. Toxicol Sci 2005;88:525-33.
- Wang AL, Wang JP, Wang H, Chen YH, Zhao L, Wang LS, et al. A dual effect of N-acetylcysteine on acute ethanol-induced liver damage in mice. Hepatol Res 2006;34:199-206.
- Laurent T, Markert M, Feihl F, Schaller MD, Perret C. Oxidant-antioxidant balance in granulocytes during ARDS: Effect of N-acetylcysteine. Chest 1996;109:163-6.
- Victor VM, Rocha M, de la Fuente M. N-acetylcysteine protects mice from lethal endotoxemia by regulating the redox state of immune cells. Free Radic Res 2003;37:919-29.
- Kwon S H, Ahn SH, Kim YK, Bae GU, Yoon JW, Hong S, et al. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J Biol Chem 2002;277:2073-80.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265-75.
- Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem 1980;106:207-12.
- Grisham MB, Johnson GG, Lancaster JR. Quantitation of nitrate and nitrite in extracellular fluids. Methods Enzymol 1996;26:237-46.
- Hsu BG, Yang FL, Lee RP, Peng TC, Harn HJ, Chen HI. N-acetylcysteine ameliorates lipopolysaccharide-induced organ damage in conscious rats. J Biomed Sci 2004;11:152-62.
- Mignon A, Rouquet N, Fabre M, Martin S, Pages JC, Dhainaut JF, et al. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med 1999;159:1308-15.
- Higuchi H, Adachi M, Miura S, Gores GJ, Ishii H. The mitochondrial permeability transition contributes to acute ethanol-induced apoptosis in rat hepatocytes. Hepatology 2001;34:320-8.
- Vairetti M, Griffini P, Pietrocola G, Richelmi P, Freitas I. Cold-induced apoptosis in isolated rat hepatocytes: protective role of glutathione. Free Radic Biol Med 2001;31:954-61.
- Li J, Quan N, Bray TM. Supplementation of N-acetylcysteine normalizes lipopolysaccharide-induced nuclear factor kappaB activation and proinflammatory cytokine production during early rehabilitation of protein malnourished mice. J Nutr 2002;132:3286-92.
- Peristeris P, Clark BD, Gatti S, Faggioni R, Mantovani A, Mengozzi M, et al. N-acetylcysteine and glutathione as inhibitors of tumor necrosis factor production. Cell Immunol 1992;140:390-9.
- Victor VM, De-la-Fuente M. N-acetylcysteine improves in vitro the function of macrophages from mice with endotoxin-induced oxidative stress. Free Radic Res 2002;36:33-45.
- Osawa Y, Nagaki M, Banno Y, Yamada Y, Imose M, Nozawa Y, et al. Possible involvement of reactive oxygen species in D-galactosamine-induced sensitization against tumor necrosis factor-alpha-induced hepatocyte apoptosis. J Cell Physiol 2001;187:374-85.
- Bergamini S, Rota C, Canali R, Staffieri M, Daneri F, Bini A, et al. N-acetylcysteine inhibits in vivo nitric oxide production by inducible nitric oxide synthase. Nitric Oxide 2001;5:349-60.