
Human C-C chemokine receptor 3 monoclonal antibody inhibits pulmonary inflammation in allergic mice1
Introduction
Asthma is an inflammatory disease in the respiratory system, which is characterized by airway inflammation hyperresponsiveness[1,2]. Clinical studies and a mouse model of asthma showed leukocytes were involved in the pathophysiological process of asthma and that eosinophils are found to play a key role in the disease due to the toxic granular proteins they secrete and the membrane products. In response to the chemokines generated locally, eosinophils from the microcirculation accumulate in the airway and result in pulmonary inflammation[3,4].
Many small molecular proteins (8–10 kDa) have been identified and characterized as chemokines by their actions on distinct subtypes of leukocytes[5,6]. Chemokines regulate immune cells by binding to 7-transmenbrane G protein-coupled receptors, including C-C chemokine receptor (CCR) 1, CCR2, and CCR3. The distribution and expression of these receptors vary in distinct cells.
CCR3 is a major chemokine receptor expressed on the surface of eosinophils and is responsible for cell activation and chemotaxis[7,8]. In addition to eosinophils, CCR3 also exist in basophils, mast cell, and Th2 cell[9,10]. In in vitro and in vivo studies, several C-C types of chemokines are shown to be activated on eosinophils in allergic inflammation. Chemokines that are selective for CCR3 include eotaxins, eotaxin-2, and eotaxin-3, whereas RANTES, MCP-3, and MCP-4 bind to other chemokine receptors other than CCR3[11]. The chemokines that contribute to aberrant eosinophil recruitment to the airways in asthma are not certain. It may be that different chemokines operate in the process of asthma by different immune mechanisms. It has been reported that the binding of an oligodeoxynucleotide to the complementary sequence of a messenger RNA, can prevent the synthesis of the encoded protein[12]. So making the blockade of a common receptor is more appealing. CCR3 have been identified and characterized from other species, such as mice, rats, guinea pigs, and non-human primates, and antibodies of CCR3 are generated to achieve the aim at this attractive biological target for therapeutic intervention[13].
The treatment of asthma has been improved by the implementation of management guidelines in recent years, with further development in the study for the mechanism of asthma. Many medicines, such as corticosteroids and theophylline have been used to control asthma as anti-inflammatory agents. However, the effects of those drugs are not satisfied in the clinical practice. Therefore, a new agent for asthma treatment is required[14,15]. For this reason, CCR3 has been more important as a significant anti-inflammatory pharmacological target, and CCR3 antagonists are currently being developed for the treatment of asthma and other allergic disorders. Administration of an anti-CCR3 monoclonal antibody (mAb) was capable of inducing a targeted reduction of eosinophils in peripheral blood. Inhibitors of CCR3 showed these agents were effective in inhibiting eosinophil recruitment in allergen models of asthma[16,17]. We also reported that antimouse CCR3 mAb inhibited airway inflammation in mice[18]. However, there is no report to evaluate the effects of the antihuman CCR3 antibody on airway inflammation, and mucus secretion in the mouse model of asthma.
Therefore, in the present study, we used the 30 amino acid synthetic peptide which is the NH2-terminal of CCR3 as the immunizing antigen and generated murine mAb against the human CCR3. Furthermore, the study was conducted on mice sensitized and challenged with ovalbumin (OVA) to determine the effects of this specific antibody. The bronchoalveolar lavage fluid (BALF), histopathology, and mucus secretion were examined. We found that the antibody we generated was specific to CCR3. The allergic mice treated with the antihuman CCR3 antibody exhibited a significant reduction of pulmonary inflammation accompanied by the alteration of cytokine in the BALF.
Materials and methods
Generation of mouse antihuman CCR3 mAb A peptide was synthesized (Wolwo Biotech, Shanghai, China), corresponding to the predicted NH2 terminus of the human CCR3 amino acid sequence (NH2-mttsldtvet fgttsyyddv gllcekadtr. Accession N
Western blot analysis of mouse antihuman CCR3 mAb Daudi cell lysates (Wolwo Biotech, China) were prepared by homogenization. Electrophoresis of secretions for CCR3 was performed on 12% (w/v) agarose gels, including 0.1% sodium dodecyl sulfate; 200 µg of the protein in each sample was solubilized in electrophoresis sample buffer. For the Western blot analysis, the membranes were blocked and the antihuman CCR3 antibody we obtained was used as the primary antibody at a dilution of 1:500. The membranes were incubated with peroxidase-conjugated antimouse IgG (Promega, Madison, WI, USA) at a dilution of 1:2000 with dilution buffer for 1 h, and the signal was developed with enhanced chemiluminescence system (ECL-Plus Amersham Life Science, Heights, IL, USA).
Mice Eight to ten-week-old male BALB/c mice weighing 20–25 g were purchased from the Laboratory Animal Center of the Medical School of Zhejiang University (Hangzhou, China. Certificate N
BALF of the lungs At d 28, BALF was performed using phosphate-buffered solution (PBS) containing 2% Fetal bovine serum (FBS). A small incision was made in the upper trachea with a needle and a catheter was inserted into the trachea through the incision. 0.5 mL 2% FBS/PBS was slowly injected into the lungs of the mice and removed repeatedly 3 times; about 1.2 mL BALF was harvested. The total BALF cellularity was determined with the use of a hemocytometer. Cytospin slides were fixed and stained by Wright–Giemsa staining. Differential cell counts by unbiased observers were based on counts of 200–300 cells using standard morphological criteria to classify individual leukocyte populations.
Pulmonary histology After BAL was performed, the lungs were inflated with 1 mL of 10% neutral-buffered formalin via a tracheotomy tube. After the instillation of the fixative, the trachea was ligated and the lung was excised and fixed in formalin for 24 h at 4 oC. These tissues were then embedded in paraffin, cut in 4 µm frontal sections, mounted onto slides, and stained with hematoxylin and eosin (HE). Periodic acid-Schiff (PAS) was used to quantitate mucus. Image-Pro Plus software (version 5.0.1, Media Cybernetics, Silver Spring, MD, USA) was used to analyze histopathology. The goblet cell hyperplasia ratio (HR) and the epithelial cell mucus occupying ratio (MOR) were used to determine airway mucus[19]: HR=(number of goblet cells/length of the airway epithelial)×100%, MOR=(area of airway epithelium staining positive for mucus/total area of airway epithelium)×100%.
Cytokine assay Interleukin-4 (IL-4) and the IFN-γ levels in the BALF were determined using the mouse IL-4 and IFN-γ ELISA kits (Jingmei BioTech, Shenzhen, China), as per the manufacturer’s instructions. The sensitivity for each cytokine was 2.0 pg/mL for IL-4 and IFN-γ.
Statistical analysis Data are expressed as mean±SEM. Statistical analysis was performed using one-way ANOVA.
Results
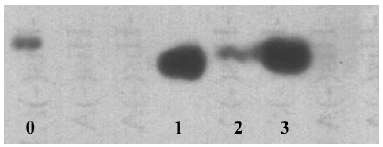
Generation and identification of the antihuman CCR3 mAb ELISA was employed to detect the affinity of the generated antibody. Antibodies from the 3 different hybridoma clones (PBND03, PBND04, and PBND05) showed high affinity with the peptide. The affinities were PBND03≥1.28×109, PBND04≥2×107, and PBND05≥3.2×108, respectively. To further identify the production of the antibody, the 3 antibodies were chosen as the primary antibodies for Western blotting. The lysates of the Daudi cell expressing CCR3 were used to determine the specificity of the generated antibody to CCR3. As shown in Figure 1, remarkable strips were observed. The antibody from the PBND03 clone was employed for further study in vivo because of its high affinity.
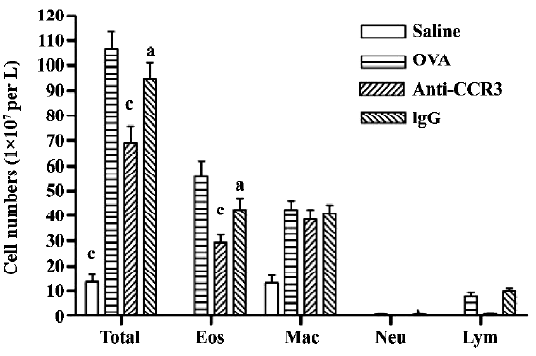
Antihuman CCR3 mAb decreases leukocytes in the BALF of allergic mice To examine the effect of antihuman CCR3 mAb on inflammatory cell accumulation responding to the allergen challenge, the BALF was recovered. The total cells of the BALF were counted and differentiated using morphological criteria. The total number of leukocytes and eosinophils in the OVA group was significantly greater than that in the saline group (P<0.01, n=4; Figure 2). A comparison of individual cell numbers per mL of BALF revealed that these increases were mainly due to increases in eosinophil populations. There were no differences in the total number of leukocytes and eosinophils between the OVA group and the IgG group. In comparison to the OVA group, the total number of leukocytes and eosinophils in the antihuman CCR3 mAb groups decreased significantly. No significant differences in neutrophils and lymphocytes were observed between the groups.
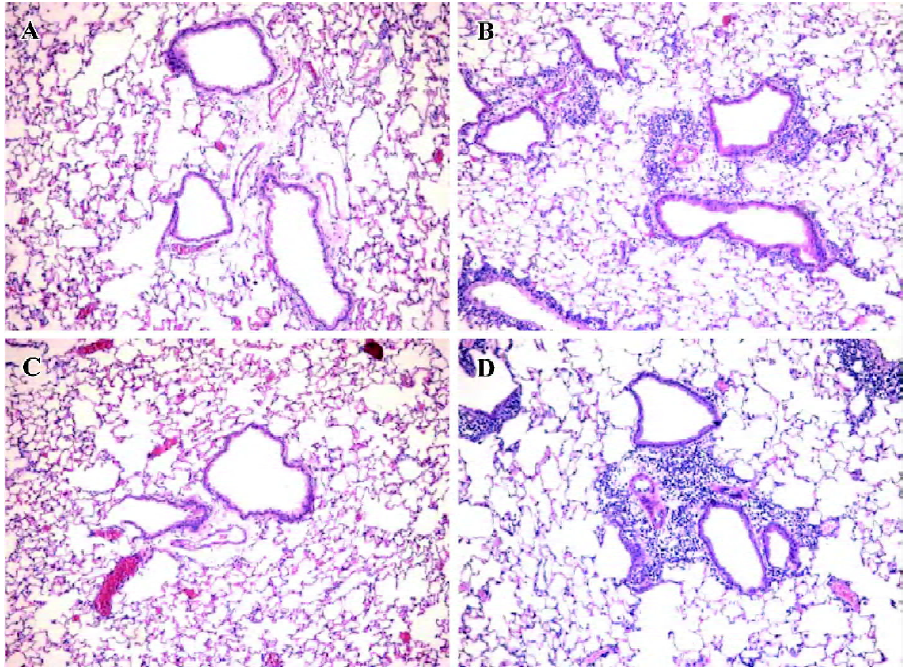
Antihuman CCR3 mAb inhibits pulmonary inflammation in allergic mice To determine if antihuman CCR3 mAb inhibits the airway inflammatory infiltration, we observed the pulmonary pathology stained with HE of the mice in all groups. Compared with the mice treated with saline, exposure to OVA resulted in the development of a characteristic peribronchial and perivascular airway tissue infiltration. In comparison to the mice in the OVA group, no obvious change of pulmonary inflammation was observed in the mice administered with non-specific IgG. However, the allergic mice administered with antihuman CCR3 mAb exhibited remarkably reduced inflammation in the lung tissues (Figure 3).
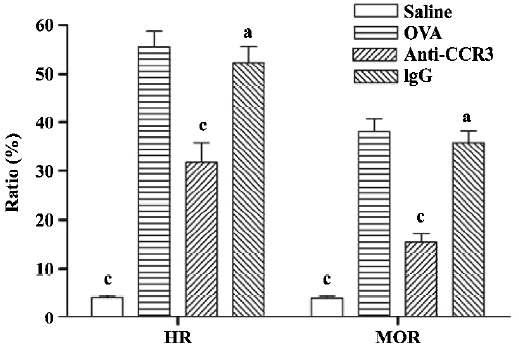
Antihuman CCR3 mAb reduces mucus overproduction of allergic mice To evaluate the inhibitory effect of antihuman CCR3 mAb on the hypersecretion of mucus in the airways, PAS stain was performed. In allergic mice, morphometric analyses of the PAS-stained lung sections revealed an obvious increase in mucus cell hypertrophy and the percentage of airway mucus, which is calculated by HR and MOR, respectively. Antihuman CCR3 mAb significantly reduced mucus secretion (Figure 4). Compared with the mice in the OVA group, the mice treated with antihuman CCR3 mAb exhibited decreased HR and MOR. No reduction of mucus hypersecretion induced by allergens was observed in the mice treated with non-specific IgG.
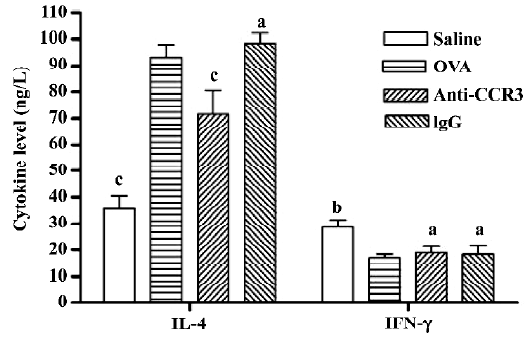
Antihuman CCR3 mAb decreases the IL-4 level in the BALF of allergic mice To determine the effect of antihuman CCR3 mAb on the pulmonary immune response in allergic mice, the concentration of IL-4 and IFN-γ in the BALF were measured. The IL-4 level in the OVA group was significantly greater than that in the saline group (P<0.01, n=4). The mice treated with antihuman CCR3 mAb showed a significant decrease of IL-4 compared to the allergic mice (P<0.01). The IFN-γ level in the OVA group was less than that in the saline group (P<0.05, n=4) and there were no differences between the antihuman CCR3 mAb groups and OVA group at the IFN-γ levels. Non-specific IgG had no effect on these cytokine levels in the allergic mice (Figure 5).
Discussion
In the present study, we described the generation of a murine monoclonal antibody against the human CCR3, which was revealed as specific for CCR3 by Western blotting. This antibody selected from the clones decreased the total number of leukocytes and eosinophils in the BALF from allergic mice. A pathology analysis found that the allergic inflammatory infiltration and mucus secretion in the lung tissues were reduced by the antibody. We also proved that the antibody decreased pulmonary inflammation induced by the allergen challenge, with the reduction of enhanced level of IL-4 in the BALF. Collectively, all of the results demonstrated the antihuman CCR3 mAb obtained in this study inhibited the airway inflammation in the mouse asthma model, which suggests that the development of potent antagonists for the chemokine receptor is viable, such as the mAb against CCR3.
A synthesized peptide corresponding to the predicted NH2 terminus of the human CCR3 amino acid sequence was chosen as the immunizing antigen to generate the antibody. To determine if the antibody generated is specific for human CCR3, we performed a Western blot analysis using the Daudi cell, which is a human B lymphoblast. CCR3 is one of receptors expressed on the surface of Daudi cells[20]. As shown in Figure 1, an obvious strip appeared in the lanes 1–3. To our knowledge, this is the first report of an antihuman CCR3 monoclonal antibody being produced that employed the NH2 terminal peptide as the antigen. Depending on the binding domain of the receptor, the monoclonal antibody against chemokine receptors are usually divided into 2 groups: one binds to the NH2 terminal region and the other recognizes the second extracellular loop. The domain that our generated antibody recognized was the NH2 terminal region, which is in agreement with a reported monoclonal antimacaque CCR3 antibody[13].
Although it is well known that eosinophil is critical in airway damage and dysfunction in asthma, the role of the cell is still questioned[21]. The results we obtained further confirm the central role of eosinophils in the pathogenesis of asthma. The infiltration of eosinophils was significantly reduced by the administration of the antibody. The reduction of eosinophils accounts for the total leukocyte decrease. The antibody also remarkably decreased the overproduction of mucus in the airways (another property of asthma) as shown by histological analyses. The anti-inflammatory activity of CCR3 blocking is supported by the existing experimental studies. It was reported that the CCR3 antibody inhibited the eosinophil chemotaxis to various chemokines[22]. Repeated antimouse CCR3 mAb treatment can partially deplete eosinophils in Nippostrongylus brasiliensis-infected lung tissue and BALF[23]. A previous study also found that the anti-CCR3 antibody could inhibit eosinophil accumulation in response to eotaxins in vivo[24]. CCR3 is the predominant chemokine receptor on human eosinophils by which many ligands bind to activate the cells. For therapeutic purposes, blocking CCR3 will be more efficient than blocking individual chemokines, such as eotaxins or RANTES.
There is a lot of evidence that supports that inflammatory cell infiltration and mucus hypersecretion are associated with Th2 cytokines that regulate immune functions in mice. IL-4 was shown to play a key role in mucus production through the recruitment of Th2 cells to the lungs and the induction of inflammation[25]. The blockage of the IL-4 receptor in mice sensitized and challenged with the antigen markedly decreased airway goblet cells metaplasia. In the absence of IL-4R alpha in recipient mice, there was no goblet cell metaplasia or mucus hypersecretion in response to OVA, even in the presence of Th2 cells and substantial eosinophilic infiltration[23]. It was reported that the expression of Th2 cytokines was unaffected as a consequence of the CCR3-mediated depletion of eosinophils in OVA-sensitized/challenged mice[26]. However, CCR3-mediated signaling can rapidly mobilize IL-4 stored in human eosinophils for release by vesicular transport to contribute to immune responses[27]. An antibody to CCR3 abrogated the eotaxin-enhanced IL-4 release in vitro[28]. The cytokine analysis in our experiments showed that the anti-CCR3 antibody significantly decreased the elevated IL-4 level in the BALF of allergic mice with reduced eosinophils. However, the antibody did not affect the level of IFN-γ in the BALF, which identical to the previous study[26]. Our results suggested that leukocytes, including eosinophil, lymphocyte and other inflammatory cells were inhibited by the CCR3 blocking pathway that contributed to the reductive synthesis of cytokine in the lungs, which leads to the decrease of airway inflammation and mucus secretion.
In conclusion, our study demonstrates that we have generated an anti-CCR3 antibody which is specific against human CCR3. The antibody could significantly inhibit the inflammation in the lungs. It is suggested that the blockade of CCR3 is an appealing therapeutical target for asthma. The present research may provide an experimental basis for the further study of this agent.
References
- O’Byrne P. Asthma pathogenesis and allergen-induced late responses. J Allergy Clin Immunol 1998;102:S85.
- Agrawal DK, Bharadwaj A. Allergic airway inflammation. Curr Allergy Asthma Rep 2005;5:142-8.
- Kumar RK, Herbert C, Thomas PS, Wollin L, Beume R, Yang M, et al. Inhibition of inflammation and remodeling by roflumilast and dexamethasone in murine chronic asthma. J Pharmacol Exp Ther 2003;307:349-55.
- Tomkinson A, Cieslewicz G, Duez C, Larson KA, Lee JJ, Gelfand EW. Temporal association between airway hyperresponsiveness and airway eosinophilia in ovalbumin-sensitized mice. Am J Respir Crit Care Med 2001;163:721-30.
- Luster AD. Chemokines – chemotactic cytokines that mediate inflammation. N Engl J Med 1998;338:436-45.
- Mackay CR. Chemokines: immunology’s high impact factors. Nat Immunol 2001;2:95-101.
- Garcia G, Godot V, Humbert M. New chemokine targets for asthma therapy. Curr Allergy Asthma Rep 2005;5:155-60.
- Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME. The eotaxin chemokines and CCR3 are fundamental regulators of allergen-inducedpulmonary eosinophilia. J Immunol 2005;175:5341-50.
- Humbles AA, Lu B, Friend DS, Okinaga S, Lora J, Al-Garawi A, et al. The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness. Proc Natl Acad Sci USA 2002;99:1479-84.
- Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science 1997;277:2005-7.
- Bisset LR, Schmid-Grendelmeier P. Chemokines and their receptors in the pathogenesis of allergic asthma: progress and perspective. Curr Opin Pulm Med 2005;11:35-42.
- Alvarez D, Wiley RE, Jordana M. Cytokine therapeutics for asthma: an appraisal of current evidence and future prospects. Curr Pharm Des 2001;7:1059-81.
- Zhang L, Soares MP, Guan Y, Matheravidathu S, Wnek R, Johnson KE, et al. Functional expression and characterization of macaque C-C chemokine receptor 3 (CCR3) and generation of potent antagonistic anti-macaque CCR3 monoclonal antibodies. J Biol Chem 2002;277:33799-810.
- Bousquet J. Global initiative for asthma (GINA) and its objectives. Clin Exp Allergy 2000;30 Suppl 1:2-5.
- Sally EW, Ronina C. Update in asthma 2005. Am J Respir Crit Care Med 2006;173:698-706.
- Ichinose M, Barnes PJ. Cytokine-directed therapy in asthma. Curr Drug Targets Inflamm Allergy 2004;3:263-9.
- Das AM, Vaddi KG, Solomon KA, Krauthauser C, Jiang X, McIntyre KW, et al. Selective inhibition of eosinophil influx into the lung by small molecule CC chemokine receptor 3 antagonists in mouse models of allergic inflammation. J Pharmacol Exp Ther 2006;318:411-7.
- Shen HH, Xu F, Zhang GS, Wang SB. CCR3 monoclonal antibody inhibits airway eosinophilic inflammation and mucus overproduction in a mouse model of asthma. Acta Pharmacol Sin 2006;27:1594-9.
- Zhang GS, Shen HH. The preventive effect of bacillus Calmette-Guerin vaccination in early life on airway inflammation and mucus production in a murine model of asthma. Zhonghua Jie He He Hu Xi Za Zhi 2005;28:17-21. Chinese..
- Abonyo BO, Alexander MS, Heiman AS. Autoregulation of CCL26 synthesis and secretion in A549 cells: a possible mechanism by which alveolar epithelial cells modulate airway inflammation. Am J Physiol Lung Cell Mol Physiol 2005;289:L478-88.
- Siegle JS, Hansbro N, Herbert C, Yang M, Foster PS, Kumar RK. Airway hyperreactivity in exacerbation of chronic asthma is independent of eosinophilic inflammation. Am J Respir Cell Mol Biol 2006;35:565-70.
- Heath H, Qin S, Rao P, Wu L, LaRosa G, Kassam N, et al. Chemokine receptor usage by human eosinophils. The importance of CCR3 demonstrated using an antagonistic monoclonal antibody. J Clin Invest 1997;99:178-84.
- Grimaldi JC, Yu NX, Grunig G, Seymour BW, Cottrez F, Robinson DS, et al. Depletion of eosinophils in mice through the use of antibodies specific for C-C chemokine receptor 3 (CCR3). J Leukoc Biol 1999;65:846-53.
- Sabroe I, Conroy DM, Gerard NP, Li Y, Collins PD, Post TW, et al. Cloning and characterization of the guinea pig eosinophil eotaxin receptor, C-C chemokine receptor-3: blockade using a monoclonal antibody in vivo. J Immunol 1998;161:6139-47.
- Steinke JW. Anti-interleukin-4 therapy. Immunol Allergy Clin North Am 2004;24:599-614.
- Kelly-Welch AE, Melo ME, Smith E, Ford AQ, Haudenschild C, Norben-Trauth N, et al. Complex role of the IL-4 receptor alpha in a murine model of airway inflammation: expression of the IL-4 receptor alpha on nonlymphoid cells of bone marrow origin contributes to severity of inflammation. J Immunol 2004;172:4545-55.
- Justice JP, Borchers MT, Crosby JR, Hines EM, Shen HH, Ochkur SI, et al. Ablation of eosinophils leads to a reduction of allergen-induced pulmonary pathology. Am J Physiol Lung Cell Mol Physiol 2003;284:L169-78.
- Bandeira-Melo C, Sugiyama K, Woods LJ, Weller PF. Cutting edge: eotaxin elicits rapid vesicular transport-mediated release of preformed IL-4 from human eosinophils. J Immunol 2001;166:4813-7.
- Devouassoux G, Metcalfe DD, Prussin C. Eotaxin potentiates antigen-dependent basophil IL-4 production. J Immunol 1999;163:2877-82.