
Dehydroevodiamine attenuates calyculin A-induced tau hyperphos-phorylation in rat brain slices1
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the aged population. It is characterized by the presence of extracellular senile plaques composed of β-amyloid and intracellular neurofibrillary tangles (NFT) constituting primarily of the abnormally hyperphosphorylated microtubule-associated protein tau[1–4]. A profound loss of basal forebrain cholinergic neurons accompanied by dysfunction of central cholinergic neurotransmission was also obvious in AD patients[5].
As the mechanism leading to the hallmark pathological alterations is still not fully understood, there is still no specific cure for the therapy of the disease. So far, the most effective pharmacological strategies to attenuate the impaired cognitive function of AD patients have been targeted at the supplementation of acetylcholine with acetylcholinesterase inhibitors[6]. However, it is just a palliative strategy aimed at the temporary improvement of the cognitive function[7]. With the progress in understanding the molecular and cellular pathophysiology of AD, pharmacological interventions aimed at modifying the development and progress of the disease have been developed. These approaches include β-amyloid immunization, treatment with secretase inhibitors, and the inhibition of tau-related neurofibrillary degeneration[6,8].
Dehydroevodiamine (DHED), one of the quinazoline alkaloids isolated from Evodia rutaecarpa Bentham, was found to be a non-competitive cholinesterase inhibitor[9]. Studies have shown that it has anti-amnesic effects in a scopolamine-induced amnesia model and can prevent impairment of learning and memory and neuronal loss in rat models of cognitive disturbance with minimal side-effects and effective dosing[9,10]. It is also demonstrated that DHED can attenuate Aβ-induced amnesia in mice, which suggests that DHED may be an ideal drug candidate for the AD-type dementia treatment[11]. The formation of neurofibrillary tangle is the recognized pathology positively correlated with the extent of neuronal loss and the degree of clinical dementia in AD[12,13]. Neurofibrillary tangles are primarily composed of the abnormally hyperphosphorylated tau. It has been found that tau hyperphosphorylation is an early event in the evolution of the disease[1–3]. Thus, we are curious as to whether DHED has an effect on the AD-like tau hyperphosphorylation and the possible underlying mechanism.
The phosphorylation of tau is strictly regulated by a panel of protein kinases and protein phosphatases (PP). Among the known phosphatases, PP-2A is the most active enzyme in dephosphorylating the abnormally hyperphosphorylated tau isolated from AD patients[14–16] or induced in the rat brain[17], and the activity of PP-2A is significantly reduced in the AD brains[18]. Therefore, in the present study, we treated the metabolically competent rat brain slice with calyculin A (CA), a potent and specific inhibitor of PP-2A and PP-1, to induce AD-like tau hyperphosphorylation, and investigated the effect of DHED on tau phosphorylation and the activity-dependent modification of PP-2A. We found that DHED could prevent the CA-induced tau hyperphosphorylation in rat brain slices and efficiently decreased the inhibitory phosphorylation of PP-2A at Tyr307.
Materials and methods
Animals Male Sprague-Dawley rats, weighing 200–240 g, were obtained from the Center of Laboratory Animals of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). All the animal experiments were performed according to the Policies on the Use of Animals and Humans in Neuroscience Research revised and approved by the Society for Neuroscience in 1995.
Chemicals and antibodies DHED was purified by HPLC from the unripe fruit of Evodia rutaecarpa Bentham with 98% purity (Kunming Biovalley Institute of Materia Medica, Kunming, China). The drug was dissolved in 5% DMSO solution and was prepared immediately prior to use[19]. CA (Discodermia calyx) and monoclonal antibody (mAb) Tau-1 against unphosphorylated tau were purchased from Chemi-con International (Temecula, CA, USA). Polyclonal antibody (pAb) R134d against total tau and pAb R123d against PP-2A catalytic subunit were gifts from Dr IQBAL and Dr GRUNDKE-IQBAL (New York State Institute for Basic Research, Staten Island, NY, USA). pAb pS262 against phosphorylated tau at Ser262 and pAb pS396 against phosphorylated tau at Ser396 were purchased from Biosource International (Camarillo, CA, USA). pAb against phosphorylated Tyr307 of PP-2A catalytic subunit (anti-p-PP2A) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). pAb anti-β-actin and mAb anti-α-tubulin were purchased from Sigma Chemical Co (St Louis, MO, USA).
Preparation and treatment of rat brain slices The rats were deeply anaesthetized and the brains were rapidly removed and cooled in oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (aCSF) containing 126 mmol/L NaCl, 3.5 mmol/L KCl, 1.2 mmol/L NaH2PO4, 1.3 mmol/L MgCl2, 2.0 mmol/L CaCl2, 11 mmol/L D-(+)-glucose, and 25 mmol/L NaHCO3 (pH 7.4) for 7–8 min at 4 °C. The forebrain was isolated and sagittally divided. Then, 400 µm thick coronal slices were sectioned with a Mcllwain Tissue Chopper (Mickle Laboratory Engineering Co Ltd, Gomshall, Surrey, England). The slices were equilibrated at room temperature for 1 h and immediately incubated at 33 °C in oxygenated aCSF either alone (control) or in the presence of DHED with different concentrations (10, 100, and 200 µmol/L, respectively). After incubation for 1 h, CA was added into the aCSF to induce tau hyperphosphorylation. After incubation for another 2 h, the brain slices were removed, washed twice and divided into 2 parts. One part was homogenized at 4 °C in the buffer containing 50 mmol/L Tris-HCl (pH 7.0) 10 mmol/L β-mercaptoe-thanol, 1.0 mmol/L EDTA, 0.1 mmol/L phenylmethylsulfonyl fluoride, 2.0 mmol/L benzamidine, 1.0 mmol/L Na3VO4, 100 mmol/L NaF, and 2.0 mg/mL each of aprotinin, leupeptin, and pepstatin A at a ratio of 1:9 (g/mL). The other part was fixed in 4% paraformaldehyde for immunohistochemistry.
Lactate dehydrogenase (LDH) activity assay The LDH released into aCSF from rat brain slices during incubation was determined colorimetrically with a commercial kit from Jiancheng Bioengineering Institute (Nanjing, China) according to the manufacturer’s protocol. The assay was carried out on 96-well microplates, and the results were read by DG3022-microplate reader (TECAN, Grodig, Salzburg, Australia) at a wavelength of 440 nm.
Western blotting The protein concentration of homo-genates from the brain slices was quantitated by the bicinchoninic acid method using protein assay reagent (Pierce, Rockford, IL, USA). Homogenates were separated by electrophoresis through 10% SDS-PAGE. After being transferred to polyvinylidene difluoride membranes (Amersham Pharmacia Biotech, Uppsala, Sweden) and probed with anti-tau and PP-2A antibodies as described above, the density of the bands was quantitatively analyzed by Kodak Digital Science 1D software (Eastman Kodak, New Haven, CT, USA).
Immunohistochemistry At the end of incubation, the slices were collected, washed with phosphate-buffered saline, and fixed in 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) at room temperature for 5 h. Then the slices were dehydrated with 70% ethanol, 80% ethanol, 95% ethanol, 100% ethanol, and xylene sequentially. The fixed slices were then embedded in paraffin and cut into 6 µm-thick serial sections. All the slides with tissue sections were dried at 37 °C for 2 d before they were immunostained with the antibodies as described above. The immunoactivities were detected using an avidin-biotin-peroxidase system and visualized with diaminobenzidine (DAB; Sigma, St Louis, MO, USA).
Statistical analysis All data were expressed as mean±SD and analyzed by using SPSS 13.0 statistical software (SPSS, Chicago, IL, USA). One-way ANOVA followed by least significant difference post-hoc tests was used to determine the differences among the groups.
Results
Effect of DHED on CA-induced tau hyperphosphorylation Data received by Western blotting (Figure 1A, upper panel) and quantitative analysis (Figure 1A, lower panel) showed a remarkably increased immunoreaction of tau to pS262 and a reduced binding of tau to Tau-1 after incubating the brain slices with CA for 2 h, suggesting hyperphos-phorylation of tau at Ser262 (pS262) and Ser198/199/202 (Tau-1, the antibody reacts with the unphosphorylated epitope of tau; Figure 1A). No obvious change in total tau (R134d) and the phosphorylated tau at Ser396 (pS396) was observed when treated with CA (Figure 1A). The administration of DHED dramatically arrested the CA-induced tau hyperphosphorylation at Tau-1 and pS262 epitopes (Figure 1A). It also decreased the basal phosphorylation level of tau at Ser396, although CA failed to induce hyperphosphorylation at this site (Figure 1A). These results suggested that DHED could efficiently attenuate CA-induced tau hyperphosphorylation at multiple AD-related sites in the rat brain slices.
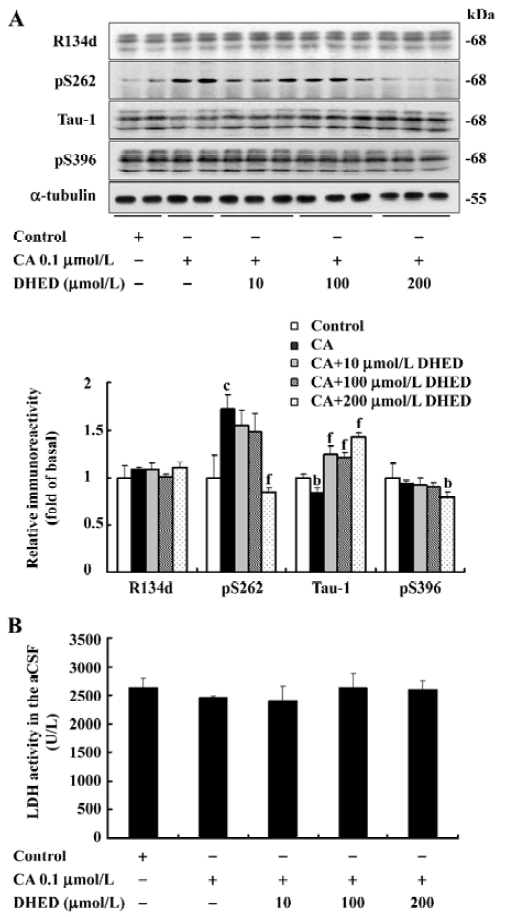
To determine the viability of the brain slices during the treatment with CA and DHED, we measured the LDH level released from the slices into the culture medium. No obvious differences were observed among the different groups (Figure 1B). These data suggested that CA at the concentration used for the study did not cause a markedly increased cell death in the cultured rat brain slices, and DHED also did not affect the cell viability.
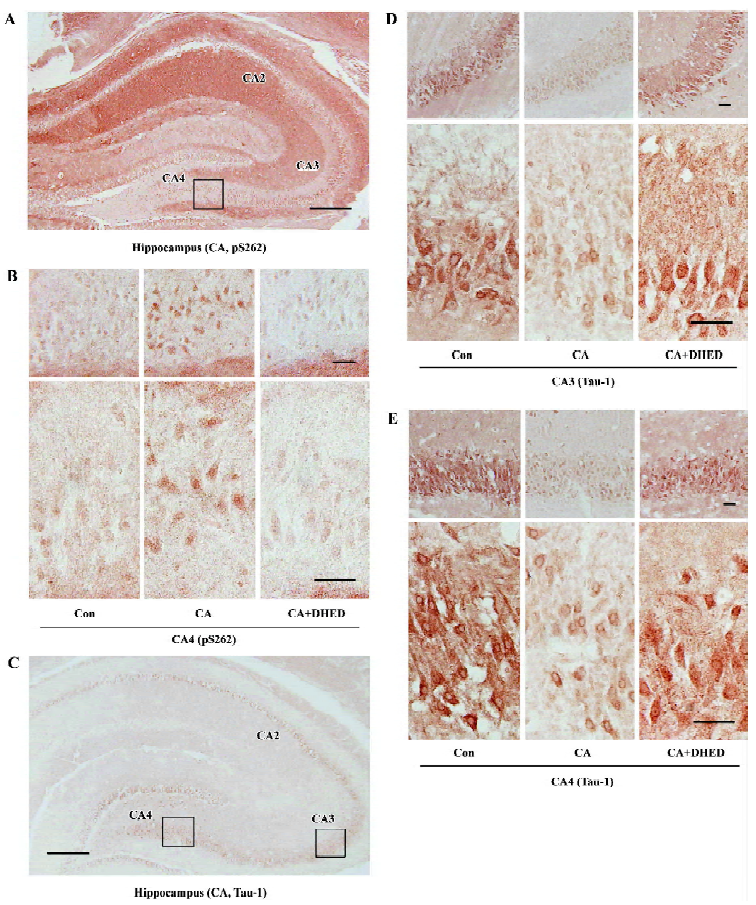
Effect of DHED and CA on the distribution of phosphorylated tau According to the results observed by Western blotting, we chose DHED at a concentration of 200 µmol/L to study the distribution of the phosphorylated tau. We further cut the fixed brain slices into 6 μm sections and measured the distribution of the phosphorylated tau and the effects of DHED by immunohistochemistry. Compared with the control group, dramatically increased pS262 staining was observed in the perinuclear of the pyramidal neurons in CA4 region of the cornu ammonis, and decreased Tau-1 staining was apparent in the cytoplasm and neural fibers of the CA3 and CA4 regions in sections treated with CA. DHED markedly reversed the hyperphosphorylation of tau at these regions (Figure 2).
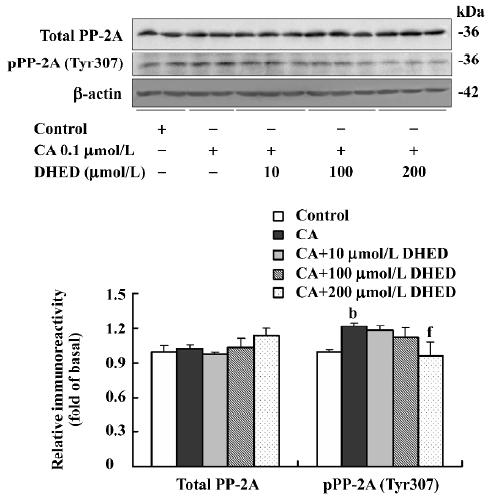
Effect of DHED and CA on Tyr307-phosphorylated PP-2A To explore the possible mechanism involved in the attenuation of DHED in the CA-induced tau hyperphosphorylation, Western blotting was used to measure the phosphorylated level of Tyr307 in the PP-2A catalytic subunit, probably contributing to the inactivation of PP-2A[20]. The phosphorylated level of Tyr307 in the PP-2A catalytic subunit was elevated in the CA-treated slices, while DHED at the concentration of 200 µmol/L efficiently decreased the inhibitory phosphorylation of PP-2A at Tyr307. No obvious difference in total PP-2A was detected among the various groups (Figure 3).
Discussion
The amount of NFT in affected neurons is directly associated with dementia symptoms in AD patients. The major component of NFT is the hyperphosphorylated tau, an early event in the evolution of AD[1–3]. Therefore, tau hyperphos-phorylation has been proposed to play a pivotal role in AD patients[21]. However, there has been no specific and effective drug to arrest tau hyperphosphorylation. In the present study, we demonstrated by Western blotting that DHED could protect against CA-induced tau hyperphosphorylation at Ser262 and Ser198/199/202 sites in rat brain slices in a dose-dependent manner. By immunohistochemistry, we further confirmed that CA could induce tau hyperphosphorylation in the hippocampus, the most critical brain region responsible for spatial learning and memory, and also the region heavily burdened with NFT in AD patients[22]. It is known that tau is hyperphosphorylated at multiple sites in the AD brains[23]. The phosphorylation of tau at Ser262 alone, a critical site located within the microtubule binding domain, dramatically decreases the binding of tau to the microtubules and thus inhibits the assembly of microtubules[24]. It may eventually lead to the collapse of cytoskeleton and neurodegeneration. Our results demonstrated that DHED could prevent tau from hyperphosphorylation in some key sites, thus, we speculate that DHED may be able to maintain the stability of microtubules and thus hold out a normal architecture of the neurons. We reported in a previous study that CA could induce hyperphosphorylation of tau at Ser396/404 by using the paired helical filament (PHF)-1 antibody in N2a cells[25]. However, we did not see increased phosphorylation of tau at Ser396 (using the pS396 antibody) when treated with CA in the rat brain slice in the present study. This discrepancy can be induced by different sources of the materials used for the studies. It also suggests that CA may predominantly affect Ser404, but not Ser396 because PHF-1 reacts with both sites, but pS396 only binds to p-Ser396. Although CA failed to induce tau hyperphosphorylation at Ser396, DHED could still decrease the basal phosphorylation level of tau at this site.
The phosphorylation of tau is strictly regulated by a panel of protein kinases and protein phosphatases. Among the latter, PP-2A is believed to be the most crucial phosphatase in regulating tau dephosphorylation[14–17]. In the AD brains, the levels of mRNA and the major heterotrimer holoenzyme, as well as the activity of PP-2A, were reduced significant-ly[18,26,27]. In vitro studies also demonstrated that PP-2A was the most active phosphatase in dephosphorylating the abnormally hyperphosphorylated and aggregated tau isolated from the AD brain and restoring the biological activity of tau in promoting microtubule assembly[14,15]. Therefore, PP-2A might be a promising target to recover normal tau function in the AD brains. However, until now, no effective agent has been reported to selectively activate PP-2A in tangle-bearing neurons. In the present study, we found that DHED treatment could ameliorate tau hyperphosphorylation, and in the meantime, it decreased the phosphorylation level of PP-2A at Tyr307, probably contributing to the inactivation of the catalytic subunit of the phosphatase. Our results strongly indicate that DHED may exert its protective effect on CA-induced tau hyperphosphorylation at least partially through antagonizing the CA-induced PP-2A inhibition. As an acetylcholinesterase inhibitor, DHED inhibits the activity of cholinesterase. In a previous study, we found that the inhibition of PP-2A by okadaic acid decreased the level of acetylcholine in the rat brain[28]. It has also been reported recently that therapeutic acetylcholinesterase inhibitors, such as donepezil and galanthamine, can prevent glutamate neurotoxicity via nicotinic acetylcholine receptors and inhibitors for a non-receptor type tyrosine kinase Fyn, and janus-activated kinase 2 suppressed the neuroprotective effect of donepezil and galanthamine[29]. These studies suggest the possible connections of cholinergic metabolism function, PP-2A activity/the tyrosine phosphorylation, and the tyrosine kinase regulation. The exact mechanism for the ameliorative effect of DHED in the CA-induced inhibitory tyrosine phosphorylation of PP-2A needs further investiga-tion. It should be noted that DHED treatments at lower concentrations (10 and 100 µmol/L) prevented CA-induced tau hyperphosphorylation at Ser198/199/202 sites (Tau-1 epitopes), but exerted no effect on the inhibitory modification of PP-2A, indicating the possible involvement of other tau kinases and phosphatases.
As DHED is easy to pass through the blood-brain barrier and has been reported to have the least side-effects and the lowest dosing among the cholinesterase inhibitors[9,10,30], in addition to the present findings, it may be a promising candidate for the drug development of AD.
Acknowledgements
We thank Dr Khalid IQBAL, Dr Inge GRUNDKE-IQBAL, Dr Cheng-xin GONG, and Dr Fei LIU at New York State Institute for Basic Research for technical support.
References
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 1986;261:6084-9.
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 1986;83:4913-7.
- Lee VM, Balin BJ, Otvos L Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991;251:675-8.
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 1985;82:4245-9.
- Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976;2:1403.
- Giacobini E. From molecular structure to Alzheimer therapy. Jpn J Pharmacol 1997;74:225-41.
- Yamada K, Toshitaka N. Therapeutic approaches to the treatment of Alzheimer’s disease. Drugs Today (Barc) 2002;38:631-7.
- Iqbal K, Grundke-Iqbal I. Inhibition of neurofibrillary degenera-tion: a promising approach to Alzheimer’s disease and other tauopathies. Curr Drug Targets 2004;5:495-502.
- Park CH, Kim SH, Choi W, Lee YJ, Kim JS, Kang SS, et al. Novel anticholinesterase and antiamnesic activities of dehydro-evodiamine, a constituent of Evodia rutaecarpa. Planta Med 1996;62:405-9.
- Park CH, Lee YJ, Lee SH, Choi SH, Kim HS, Jeong SJ, et al. Dehydroevodiamine·HCl prevents impairment of learning and memory and neuronal loss in rat models of cognitive disturbance. J Neurochem 2000;74:244-53.
- Wang HH, Chou CJ, Liao JF, Chen CF. Dehydroevodiamine attenuates beta-amyloid peptide-induced amnesia in mice. Eur J Pharmacol 2001;413:221-5.
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hugman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992;42:631-9.
- Riley KP, Snowdon DA, Markesbery WR. Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann Neurol 2002;51:567-77.
- Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem 1995;270:4854-60.
- Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res 1996;38:200-8.
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 2005;22:1942-50.
- Gong CX, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 2000;275:5535-44.
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem 1995;65:732-8.
- Peng JH, Zhang CE, Wei W, Hong XP, Pan XP, Wang JZ. Dehydroevodiamine attenuates tau hyperphosphorylation and memory deficit induced by activation of glycogen synthase kinase-3 in rats. Neuropharmacology 2007;52:1521-7.
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992;257:1261-4.
- Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Dysregulation of protein phosphorylation/dephosphorylation in Alzheimer’s disease: a therapeutic target. J Biomed Biotechnol 2006; 2006: 31825.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239-59.
- Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer’s disease. J Neural Transm 2005;112:813-38.
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993;11:153-63.
- Li XC, Wang ZF, Zhang JX, Wang Q, Wang JZ. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur J Pharmacol 2005;510:25-30.
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, et al. Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol 2004;63:287-301.
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol 2001;168:402-12.
- Tian Q, Lin ZQ, Wang XC, Chen J, Wang Q, Gong CX, et al. Injection of okadaic acid into the Meynert nucleus basalis of rat brain induces decreased acetylcholine level and spatial memory deficit. Neuroscience 2004;126:277-84.
- Takada-Takatori Y, Kume T, Sugimoto M, Katsuki H, Sugimoto H, Akaike A. Acetylcholinesterase inhibitors used in treatment of Alzheimer’s disease prevent glutamate neurotoxicity via nicoginic acetylcholine receptors and phosphatidylinositol 3-kinase cascade. Neuropharmacology 2006;51:474-86.
- Ahn SH, Jeon SH, Tsuruo T, Shim CK, Chung SJ. Pharmacokinetic characterization of dehydroevodiamine in the rat brain. J Pharm Sci 2004;93:283-92.