
Green tea polyphenol epigallocatechin-3-gallate suppresses rat hepatic stellate cell invasion by inhibition of MMP-2 expression and its activation1
Introduction
Hepatic fibrosis is a dynamic and sophisticatedly regulated wound healing response to chronic hepatocellular injury. This fibrotic process results from the accumulation of extracellular matrix (ECM) including collagen, proteoglycan and adhesive glycoproteins. Hepatic stellate cells (HSC) are recognized as the primary cellular source of matrix components in chronic liver disease, and therefore play a critical role in the development and maintenance of liver fibrosis[1]. In the healthy liver, HSC express a quiescent phenotype and are responsible for the storage of vitamin A located within the subendothelial space of Disse[2]. During fibrosis, HSC become activated and transform into proliferating fibroblast-like cells which display increased proliferation and migration, enhanced expression of matrix protein, increased production of matrix metalloproteinases (MMP) and tissue inhibitors of metalloproteinases (TIMP), all of which lead to replacement by interstitial collagen or scar matrix[3].
Many studies have demonstrated that in hepatic fibrosis progression, the expression of MMP involved in fibrillar collagen degradation (eg MMP-1 in humans and MMP-13 in rats) is very limited, whereas the expression of MMP-2 is markedly increased. Evidence from experimental and clinical studies indicates that MMP-2 expression and upregulated activity is one of the major causes of liver fibrosis[4–6]. MMP-2 is constitutively expressed and secreted as a latent zymogen (pro-MMP-2) which is activated by a membrane-linked process mediated by MT1-MMP and TIMP-2[7,8]. Once activated, MMP-2 is able to degrade the normal subendothelial matrix, hastening its replacement by fibrillar collagen. The presence of this newly-formed collagen lattice can further promote HSC activation[5].
Although the underlying mechanisms remain incompletely understood, accumulating evidence indicates that the migration of activated HSC from the sinusoidal wall and subsequent movement into regions of injury is a key event for the development of fibrosis. To migrate, HSC must degrade the subendothelial matrix that is rich in collagen IV, the principal substrate for MMP-2[9].
Experimental and clinical results indicate that oxidative stress plays critical roles in the activation of HSC and hepatic fibrogenesis[10]. MMP-2 activation is a critical pathophysiological mechanism relating to the survival of HSC, mediated during the response to oxidative stress[11–13]. Reducing oxidative stress by antioxidants could be a potential and effective therapeutic strategy for prevention and treatment of hepatic fibrosis.
Recently, a major polyphenol of green tea, epigallocate-chin-3-gallate (EGCG), was implicated as the main active ingredient. EGCG is a potent antioxidant that has attracted considerable attention for its role in preventing oxidative stress-related diseases including cancers, cardiovascular diseases and fibrosis[14–16]. The inhibiting effects of EGCG on fibroblasts and vascular smooth muscle cell MMP-2 expression and activation have been recently investigated[15,17]. However, to our knowledge, no studies have been done to investigate these effects on HSC.
To gain further insight into the effects of EGCG on the suppression of hepatic fibrosis, the present study investigated the effects of EGCG on MMP-2 expression and activation, as well as the in vitro migration and invasion of rat HSC.
Materials and methods
Reagents EGCG (purity >95%) was purchased from Sigma-Aldrich Chemical Co (St Louis, MO, USA); MMP-2 inhibitor I (OA-Hy) was obtained from Calbiochem (La Jolla, CA, USA); antibodies against MMP-2 and TIMP-2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and antibodies against MT1-MMP were obtained from Chemicon (Temecula, CA, USA).
Culture of HSC Rat HSC were kindly donated by Professor Shi-gang XIONG (Department of Pathology, Keck School of Medicine, University of Southern California, USA). They were cultured in Dulbecco’s minimal essential medium (DMEM; Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Bio-Whittaker, Walkersville, MD, USA), 100 IU/mL penicillin and 100 µg/mL streptomycin. Cultures were incubated at 37 oC in a humidified atmosphere of 5% CO2 and the medium was changed twice a week. Cell viability was tested by trypan blue exclusion. All experiments were performed with HSC obtained after 6–15 passages following exposure to serum-free culture medium containing 0.1% bovine serum albumin for 24 h.
Gelatin zymography HSC (5.5×104/well) in 6-well plastic dishes were cultured with ConA (20 µg/mL) in the presence or absence of EGCG at the indicated concentration for 24 h. MMP-2 activity in the conditioned medium of cultured HSC was analyzed by substrate-gel electrophoresis (zymography) using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 7.5%) containing 0.1% gelatin (Sigma-Aldrich). Equal volumes of samples of conditioned cell culture medium were mixed with Laemmli buffer under non-reducing conditions, loaded onto the gel and separated by electrophoresis. After electrophoresis, SDS was removed by soaking the gels 3 times for 30 min at room temperature in buffer (50 mmol/L Tris–HCl, pH 8.0, 5 mmol/L CaCl2, 0.02% NaN3 and 2.5% Triton X-100) and incubated for 24 h at 37 oC with the same buffer lacking Triton X-100. Gels were then stained with 0.1% Coomassie Brilliant Blue R-250 and de-stained until clear bands became evident. Quantitative results of the assays were obtained by densitometry.
RT-PCR HSC (5.5×104/well) in 6-well plastic dishes were cultured with ConA (20 µg/mL) in the presence or absence of EGCG at the indicated concentration for 24 h. Total RNA was extracted by the use of TRIzol reagent (Sigma-Aldrich) according to the manufacturer’s instructions. Reverse transcription with oligo (dT) priming was used to generate cDNA from total RNA (2 µg) extracts. The synthesized cDNA for MMP-2, TIMP-2, MT1-MMP and β-actin were amplified using specific sets of primers. Rat MMP-2 forward primer was 5' GCT GAT ACT GAC ACT GGT ACT G 3' and reverse primer was 5' CAA TCT TTT CTG GGA GCT C 3'[18]. Rat MT1-MMP (MMP-14) forward primer was 5' GTA CTA CCG CTT CAA TGA GG 3' and reverse primer was 5' CAC TGC CAG TAC CAG GAG 3'[19]. Rat TIMP-2 forward primer was 5' ATT TAT CTA CAC GGC CCC 3' and reverse primer was 5' CAA GAA CCA TCA CTT CTC TTG 3'[20]. β-actin primer was designed based on published cDNA sequences. The forward primer was 5' TGG GAC GAT ATG GAG AAG AT 3' and the reverse primer was 5' ATT GCC GAT AGT GAT GAC CT 3'. Each PCR mixture contained the appropriate set of forward and reverse primers (0.2 µmol/L), each dNTP at 0.25 mmol/L, 1.25 U Taq polymerase, and 2.5 mmol/L MgCl2 in a PCR buffer. The PCR procedure consisted of 28 cycles of denaturation at 95 oC for 1 min, annealing at 58 oC for 1 min and extension at 72 oC for 1 min, with initial denaturation of sample cDNA at 95 oC for 3 min and an additional extension period of 10 min after the last cycle. The PCR products were subjected to 1.5% agarose gel electrophoresis, staining with ethidium bromide and quantitation by densitometry using the Image Master VDS system and associated software (Pfizer, NY, USA).
Western blotting Cells were washed with cold PBS and lysed by the addition of a lysis buffer containing 1% Nonidet P-40, 50 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 0.1% SDS, and protease inhibitor cocktail (Boehringer Mannhein, Lewes, UK.) for 20 min at 4 oC. Insoluble materials were removed by centrifugation at 15 000 g for 15 min at 4 oC. The supernatant was saved and the protein concentration was determined using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA). Cell extracts (50 µg/lane) were separated via 10% gel electrophoresis and electroblotted onto polyvinylidene fluoride (PVDF) membranes. Nonspecific binding sites were blocked by incubating PVDF membranes for 1 h in phosphate-buffered saline containing 5% low-fat dry milk. Membranes were probed with primary antibodies overnight at 4 oC, followed by the application of a secondary horseradish peroxidase-conjugated second antibody. Blots were developed using an enhanced chemiluminescence detection system (ECL, Amersham Pharmacia Biotech, Piscataway, NJ, USA) according to the manufacturer’s instructions.
Cellular MT1-MMP activity assay HSC (1×104/well) in 24-well plastic dishes were cultured with ConA (20 µg/mL) in the presence or absence of EGCG at the indicated concentration for 24 h. MT1-MMP activity was determined with the use of a commercial kit (Amersham) according to the manufac-turer’s instructions. Briefly, MT1-MMP was extracted from cultured HSC by extraction buffer and its activity detected through activation of the modified pro detection enzyme and the subsequent cleavage of its chromogenic peptide subs-trate. The resultant color was read at 405 nm on a microplate spectrophotometer. The concentration of active MT1-MMP in a sample was determined by interpolation from a standard curve.
Wound healing assay To study the effects of EGCG on HSC migration, a wound healing assay was performed following standard methods[21]. Briefly, HSC were seeded in 6-well plates in DMEM containing 10% FBS and grown until 90% confluent. Cells were then serum starved for 24 h, and a linear wound was created in the confluent monolayer using a 200 µL pipette tip. Cells were then washed with PBS and diluted in DMEM containing 1% FBS. For the evaluation of ‘wound closure’ in the different experimental conditions, 5 randomly selected points along each wound were marked and the horizontal distance of migrating cells from the initial wound was measured after wounding. The cell migration distance was determined by measuring the width of the wound divided by 2 and by subtracting this value from the initial half-width of the wound.
Cell invasion assay The invasion assay was performed with Transwell 24-well tissue-culture plates (Costar; Corning, NY, USA) composed of a polycarbonate membrane containing 8 µm pores. The lower and upper parts of the membrane were coated with 10 µL of type I collagen (0.5 mg/mL) and 20 µL Matrigel (1 mg/mL; Collaborative Research Inc, Bedford, MA). HSC were seeded on the inner chamber of the Transwell unit at 2×104 cells in 100 µL serum free medium with or without ConA (20 µg/mL) supplemented with EGCG or OA-Hy (100 µmol/L). The inner chamber was placed into the outer chamber, which contained 600 µL of DMEM containing 0.1% FBS and was incubated for 24 h at 37 oC in a CO2 incubator. At the end of the incubation, the cells on the upper surface of the membrane were completely removed by wiping with a cotton swab. The cells that had invaded the lower surface of the membrane were fixed with methanol and stained with hematoxylin, and photographed. Cells from various areas of the lower surface were counted using a computerized video image analyzing system (Leica Quantimet Q500MC). Each assay was undertaken in triplicate.
Statistical analyses Results were expressed as the mean±SE of at least 3 separate experiments. Results were analyzed by one-way analysis of variance (ANOVA) followed by the Student-Neumann-Keuls test. Differences with P values of <0.05 were considered significant.
Results
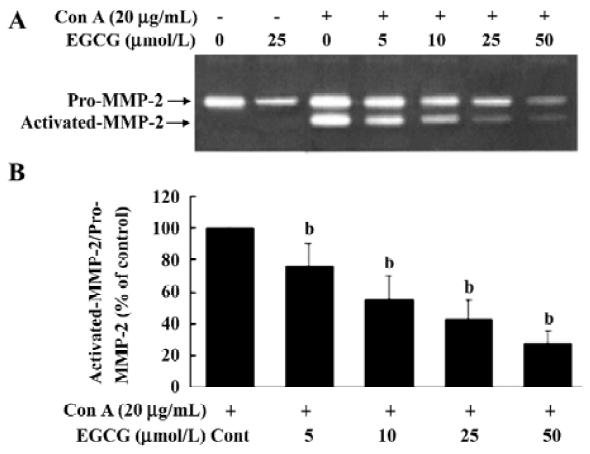
EGCG influence on ConA-induced activation of MMP-2 MMP-2 is secreted as inactive zymogen pro-MMP-2 by HSC and ConA induces pro-MMP-2 activation[22,23]. To investigate the inhibitory effects of EGCG on MMP-2 activation, the release of MMP-2 in the medium was assessed by gelatin zymography. Zymography of culture media of unstimul-ated HSC revealed a gelatinolytic band of 72 kDa that represented pro-MMP-2 (Figure 1A). After addition of Con-A to HSC and culture for 24 h, pro-MMP-2 levels were increased. In addition, less intense bands of 66 kDa were present in zymograms. The 66 kDa band corresponded to the activated form of MMP-2. EGCG at 5, 10, 25, or 50 µmol/L significantly reduced the ConA-induced activation of MMP-2 by 23.5%±6.1%, 59.4%±8.7%, 74.7%±13.5%, and 84.5%±10.6%, respectively (Figure 1B), suggesting that EGCG inhibited pro-MMP-2 activation in a dose-dependent manner. Moreover, the basal secretion of pro-MMP-2 was significantly reduced with EGCG treatment.
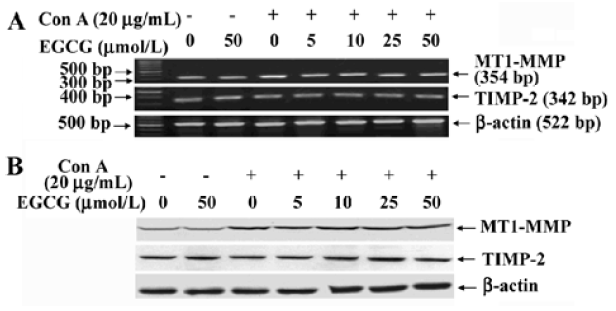
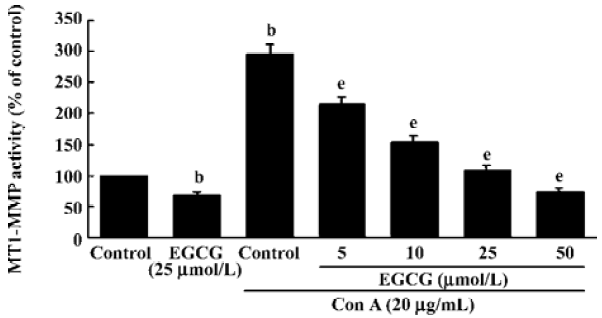
Inhibition of MT1-MMP activity by EGCG To examine the influence of EGCG on the expression of components of the MMP-2 activation complex, we assessed MT1-MMP and TIMP-2 expression in ConA-stimulated HSC treated with or without the indicated concentrations of EGCG. The results showed that the expression of TIMP-2 mRNA and protein were not affected by ConA (Figure 2A). EGCG had no influence on the level of TIMP-2, as compared with the HSC cultured without EGCG. Previous studies have suggested that the ConA-induced activation of pro-MMP-2 on the cell surface is mediated by MT1-MMP[22]. Therefore, the possibility that EGCG could prevent ConA-induced MMP-2 activation by inhibiting MT1-MMP expression in HSC was further investigated. Expression of MT1-MMP mRNA and protein was increased by ConA-stimulation. However, EGCG had no influence on the level of MT1-MMP with or without ConA (Figure 2A,2B). However, the cell-associated MT1-MMP activity was strongly inhibited by treatment of HSC with EGCG in a dose-dependent manner (Figure 3). These results indicated that EGCG reduced ConA-induced MMP-2 activation through inhibition of MT1-MMP activity.
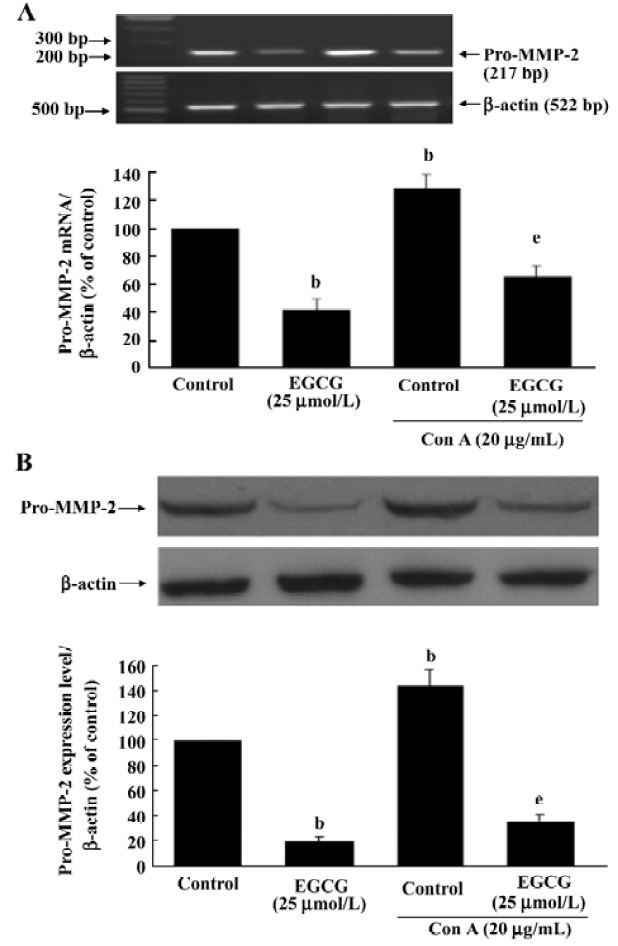
Inhibition of pro-MMP-2 expression by EGCG The possibility of EGCG affecting the amount of pro-MMP-2 expression in HSC was assessed by RT-PCR and Western blot analyses. Serum-starved HSC were cultured with or without ConA (20 µg/mL) in the presence or absence of EGCG for 24 h. The expression of MMP-2 mRNA and protein was upregulated by ConA stimulation. EGCG alone or in the presence of ConA markedly reduced MMP-2 mRNA after 24-h incubation period (Figure 4A; P<0.05). Subsequently, as shown by Western blotting, the quantity of pro-MMP-2 protein could also be significantly reduced by EGCG treatment (Figure 4B; P<0.05).
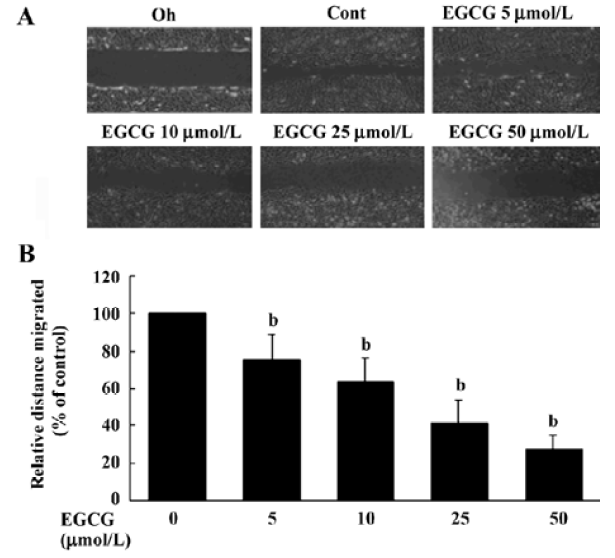
EGCG influence on cell migration in the wound healing assay The wound healing migration assay is an established and widely used procedure that allows an examination of cell migration in response to an artificial wound produced on a cell monolayer. Incubation of HSC with 1% FBS produced a marked cell migration in the wound area 24 h after wounding, whereas wounds treated with EGCG showed dose-dependent delays in wound healing under the same conditions (Figure 5A). The percentage inhibition of wound closure was evaluated on migration distance. As shown in Figure 5A, compared with 1% FBS stimulation, EGCG at 5, 10, 25, or 50 µmol/L in the media significantly reduced cell migration distance by 24.5%±7.2%, 37.4%±9.3%, 58.7%±11.8%, and 72.1%±10.3%, respectively (Figure 5B). These results indicate that EGCG inhibited the motility of HSC in vitro.
EGCG effects on HSC invasiveness To evaluate whether ConA-induced invasion of HSC could be affected by EGCG treatment, the influence of EGCG on the ability of HSC to invade through reconstituted basement membranes (Matrigel) was tested. Matrigel is a commercial product extracted from a mouse sarcoma rich in extracellular matrix protein. The major component is laminin, followed by collagen IV and heparan sulfate proteoglycans. As shown in Figure 6, ConA was found to stimulate matrix invasion of HSC, but this response was markedly reduced by EGCG, whereas EGCG alone did not affect cell invasion.
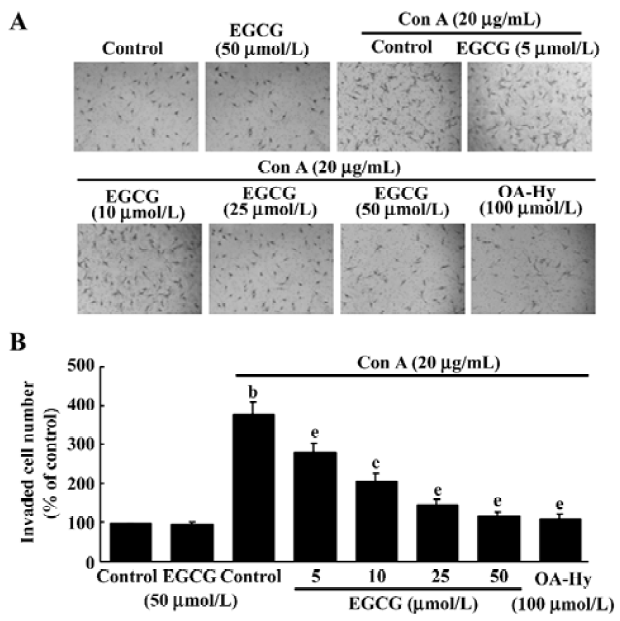
To confirm the role of MMP-2 in the regulation of invasion of HSC, we added the specific MMP-2 inhibitor I (OA-Hy) which completely abrogated the effects of ConA treatment on the invasion of HSC (Figure 6).
Discussion
MMP-2 expression and upregulated activity are one of the major causes of hepatic fibrosis. Increased MMP-2 activity is believed to be associated with an increase in destruction of the normal liver architecture, promoting its replacement by interstitial collagen[4,6]. It is therefore possible that reducing MMP-2 activity of HSC could be a potent therapeutic means for preventing hepatic fibrogenesis. In the present report, we reveal that EGCG is a strong inhibitor of the expression of pro-MMP-2 mRNA and protein, and more importantly, of the activation of the secreted MMP-2 in HSC.
MMP-2 is secreted as an inactive proenzyme. Several reports have demonstrated that activation of pro-MMP-2 at the cell surface through a trimolecular complex that includes MT1-MMP and tissue inhibitor metalloprotease-2 (TIMP-2). MT1-MMP complexed with TIMP-2 serves as a cell surface receptor for pro-MMP-2 by promoting its pericellular proteolysis and consequent activation[7,24]. However, excess TIMP-2 was observed to specifically inhibit both MMP-2 gelatinolytic activity and pro–MMP-2 activation by MT1-MMP[25]. The present findings reveal that EGCG strongly reduces the formation of active MMP-2 in response to ConA in HSC, in which the levels of activation of pro-MMP-2 were significantly inhibited by EGCG even at a very low dosage (5 µmol/L). ConA-induced activation of pro-MMP-2 is attributed to MT1-MMP[22]. Our results demonstrate that ConA can increase the expression level of MT1-MMP mRNA and protein. However, neither mRNA nor protein expression of MT1-MMP are affected with EGCG treatment in HSC. Although TIMP-2 is considered to be an inhibitor of MMP-2, expression of this inhibitor was unchanged with EGCG treatment. In contrast, EGCG markedly inhibits MT1-MMP activity in a dose-dependent manner. These results suggest that the prevention of MMP-2 activation by EGCG is likely to be mediated by the inhibition of MT1-MMP activity. A similar result in human vascular smooth muscle cells was reported[15], but the mechanism remains unknown. Previous studies have demonstrated that MMP-2 expression requires the activation of transcription factor NF-κB[26]. Since EGCG prevents the activation of NF-κB[27], its effects on MMP-2 expression is quite likely achieved through this inhibition.
In response to liver injuries, HSC are activated and accumulate at the site of injury, where they produce an extracellular matrix leading to characteristic patterns of collagen deposition[28]. Migration of resident HSC in the space of Disse is considered important for progression of liver fibrosis because it accounts for increased numbers of activated HSC in areas of injury[29]. Blockade of HSC migration may represent a potential strategy for therapy of liver fibrosis[30]. Based on insights from these and other recent works, we were prompted to examine the inhibitory effects of EGCG on HSC migration and invasion. In the current study, we examined the effects of EGCG on HSC migration measured by wound healing assay. Our results reveal that, in the presence of EGCG, HSC display a dose-dependent inhibition of wound healing. This observation suggests that EGCG is capable of inhibiting the motility of HSC. The fact that this function of EGCG dose not occur in vascular smooth muscle cells[31] suggests a differential response to EGCG based on cell type. Clearly, additional experiments are necessary to elucidate effects of this antioxidant on cytoskeleton and membrane activity required for HSC migration, as well as the involved mediators and signal transduction pathway. Finally, and more importantly, in our Transwell invasion model, we observed that ConA-induced HSC invasion was inhibited by EGCG as efficiently as the MMP-2 specific inhibitor OA-Hy. This striking effect further supports the potential role of EGCG in control of matrix degradation.
The process of HSC invasion requires the active degradation of environmental barriers including components of the basement membrane and extracellular matrix[32]. Activated HSC increase their expression and secretion of the active form of the MMP-2, which is known to be crucial in the invasion process. Recently, the central role of MMP-2 in the proliferation and invasiveness of HSC was underlined in a study that documented that collagen type I activation of the discoidin domain receptor 2 increases HSC proliferation and invasion, a phenomenon that directly reflects the increased expression of active MMP-2[33]. Furthermore, both the broad spectrum MMP inhibitor GM6001 and OA-Hy completely abrogate the oxidative stress induced proliferation and invasiveness of HSC[11]. Therefore, it is clear that the antagonistic effects of EGCG on MMP-2 expression and activation are involved in the inhibition of HSC invasion.
Oxidative stress may be a common factor in chronic liver diseases of different etiologies[10]. Many agents have been proposed for the prevention and treatment of fibrosis. However, there is no established therapy for the resolution of hepatic fibrosis. New clinical approaches need to be developed to improve the efficiency of current treatments. The antioxidant potential of EGCG is far greater than that of vitamin E and/or C[34], which might allow it to succeed where other antioxidants have failed in preventing hepatic fibrosis. In vitro studies have shown that EGCG exerts anti-fibrogenic effects by decreasing the synthesis of type I collagen, reducing cell proliferation, and by inducing apoptosis on cultured HSC[35–37]. Taken together, these findings support the suggestion that EGCG is a promising agent for the treatment of hepatic fibrosis. Whether EGCG has the same effects in vivo needs to be further investigated.
In summary, our results demonstrate that EGCG strongly inhibits pro-MMP-2 expression as well as the conversion of pro-MMP-2 into its activated form through the direct inhibition of MT1-MMP activity in cultured HSC. In addition, EGCG can inhibit HSC migration or invasion through the reconstituted basement membrane. These observations suggest a possible role for EGCG in the treatment and prevention of hepatic fibrosis.
References
- Friedman SL. Liver fibrosis-from bench to bedside. J Hepatol 2003;38:S38-53.
- Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis 2001;21:311-35.
- Friedman SL, Bansal MB. Reversal of hepatic fibrosis –– fact or fantasy? Hepatology 2006;43:S82-8.
- Preaux AM, Mallat A, Nhieu JT, D’Ortho MP, Hembry RM, Mavier P. Matrix metalloproteinase-2 activation in human hepatic fibrosis by cell-matrix interaction. Hepatology 1999;30:944-50.
- Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis 2001;21:373-84.
- Gaca MD, Zhou X, Issa R, Kiriella K, Iredale JP, Benyon RC. Basement membrane-like matrix inhibits proliferation and collagen synthesis by activated rat hepatic stellate cells: evidence for matrix-dependent deactivation of stellate cells. Matrix Biol 2003;22:229-39.
- Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res 2003;92:827-39.
- Fridman R. Surface association of secreted matrix metalloproteinases. Curr Top Dev Biol 2003;54:75-100.
- Preaux AM, Mallat A, Nhieu JT, D’Ortho MP, Hembry RM, Mavier P. Matrix metalloproteinase-2 activation in human hepatic fibrosis by cell-matrix interaction. Hepatology 1999;30:944-50.
- Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol 2001;35:297-306.
- Galli A, Svegliati-Baroni G, Ceni E, Milani S, Ridolfi F, Salzano R, et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology 2005;41:1074-84.
- Migita K, Maeda Y, Abiru S, Komori A, Yokoyama T, Takii Y, et al. Peroxynitrite-mediated matrix metalloproteinase-2 activation in human hepatic stellate cells. FEBS Lett 2005;579:3119-25.
- Gardi C, Arezzini B, Fortino V, Comporti M. Effect of free iron on collagen synthesis, cell proliferation and MMP-2 expression in rat hepatic stellate cells. Biochem Pharmacol 2002;64:1139-45.
- Hendrich AB. Flavonoid-membrane interactions: possible consequences for biological effects of some polyphenolic compounds. Acta Pharmacol Sin 2006;27:27-40.
- El Bedoui J, Oak MH, Anglard P, Schini-Kerth VB. Catechins prevent vascular smooth muscle cell invasion by inhibiting MT1-MMP activity and MMP-2 expression. Cardiovasc Res 2005;67:317-25.
- Dona M, Dell’Aica I, Calabrese F, Benelli R, Morini M, Albini A, et al. Neutrophil restraint by green tea: inhibition of inflammation, associated angiogenesis, and pulmonary fibrosis. J Immunol 2003;170:4335-41.
- Hung CF, Huang TF, Chiang HS, Wu WB. (-)-Epigallocatechin-3-gallate, a polyphenolic compound from green tea, inhibits fibroblast adhesion and migration through multiple mechanisms. J Cell Biochem 2005;96:183-97.
- Mart HP, McNeil L, Davies M, Martin J, Lovett DH. Homology cloning of rat 72 kDa type IV collagenase: cytokine and second-messenger inducibility in glomerular mesangial cells. Biochem J 1993;15:441-6.
- Okada A, Bellocq JP, Rouyer N, Chenard MP, Rio MC, Chambon P, et al. Membrane-type matrix metalloproteinase (MT-MMP) gene is expressed in stromal cells of human colon, breast, and head and neck carcinomas. Proc Natl Acad Sci USA 1995;28:2730-4.
- Cook F, Burke JS, Bergman KD, Quinn CO, Jeffrey JJ, Partridge NC. Cloning and regulation of rat tissue inhibitor of metall-oproteinases-2 in osteoblastic cells. Arch Biochem Biophys 1994;311:313-20.
- Valster A, Tran NL, Nakada M, Berens ME, Chan AY, Symons M. Cell migration and invasion assays. Methods 2005;37:208-15.
- Theret N, Lehti K, Musso O, Clement B. MMP2 activation by collagen I and concanavalin A in cultured human hepatic stellate cells. Hepatology 1999;30:462-8.
- Benyon RC, Hovell CJ, Da Gaca M, Jones EH, Iredale JP, Arthur MJ. Progelatinase A is produced and activated by rat hepatic stellate cells and promotes their proliferation. Hepatology 1999;30:977-86.
- Maquoi E, Frankenne F, Baramova E, Munau C, Sounni NE, Remacle A, et al. Membrane type 1 matrix metalloproteinase-associated degradation of tissue inhibitor of metalloproteinase 2 in human tumor cell lines. J Biol Chem 2000;14:11368-78.
- Zhao H, Bernardo MM, Osenkowski P, Sohail A, Pei D, Nagase H, et al. Differential inhibition of membrane type 3 (MT3)-matrix metalloproteinase (MMP) and MT1-MMP by tissue inhibitor of metalloproteinase (TIMP)-2 and TIMP-3 rgulates pro-MMP-2 activation. J Biol Chem 2004;279:8592-601.
- Li L, Mamputu JC, Wiernsperger N, Renier G. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: inhibitory effect of metformin. Diabetes 2005;54:2227-34.
- Chen A, Zhang L, Xu J, Tang J. The antioxidant (-)-epigallo-catechin-3-gallate inhibits activated hepatic stellate cell growth and suppresses acetaldehyde-induced gene expression. Biochem J 2002;368:695-704.
- Johnson SJ, Hines JE, Burt AD. Macrophage and perisinusoidal cell kinetics in acute liver injury. J Pathol 1992;166:351-8.
- Marra F, Romanelli RG, Giannini C, Failli P, Pastacaldi S, Arrighi MC, et al. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology 1999;29:140-8.
- Kikuchi S, Griffin CT, Wang SS, Bissell DM. Role of CD44 in epithelial wound repair: migration of rat hepatic stellate cells utilizes hyaluronic acid and CD44v6. J Biol Chem 2005;15:15398-404.
- Maeda K, Kuzuya M, Cheng XW, Asai T, Kanda S, Tamaya-Mori N, et al. Green tea catechins inhibit the cultured smooth muscle cell invasion through the basement barrier. Atherosclerosis 2003;166:23-30.
- Takahara T, Fukui K, Yata Y, Jin B, Zhang LP, Nambu S, et al. Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers. Hepatology 1997;26:1521-9.
- Olaso E, Ikeda K, Eng FJ, Xu L, Wang LH, Lin HC, et al. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest 2001;108:1369-78.
- Rice-Evans C. Implications of the mechanisms of action of tea polyphenols as antioxidants in vitro for chemoprevention in humans. Proc Soc Exp Biol Med 1999;220:262-6.
- Nakamuta M, Higashi N, Kohjima M, Fukushima M, Ohta S, Kotoh K, et al. Epigallocatechin-3-gallate, a polyphenol component of green tea, suppresses both collagen production and collagenase activity in hepatic stellate cells. Int J Mol Med 2005;16:677-81.
- Higashi N, Kohjima M, Fukushima M. Epigallocatechin-3-gallate, a green-tea polyphenol, suppresses Rho signaling in TWNT-4 human hepatic stellate cells. J Lab Clin Med 2005;145:316-22.
- Sakata R, Ueno T, Nakamura T, Sakamoto M, Torimura T, Sata M. Green tea polyphenol epigallocatechin-3-gallate inhibits platelet-derived growth factor-induced proliferation of human hepatic stellate cell line LI90. J Hepatol 2004;40:52-9.