
CCR3 monoclonal antibody inhibits airway eosinophilic inflammation and mucus overproduction in a mouse model of asthma1
Introduction
Allergic asthma is a chronic disease characterized by airway hyperresponsiveness (AHR), airway inflammation, and reversible airway obstruction. Since the mid-1980s, there has been a wealth of information documenting a potential pathogenic and destructive role for eosinophils in asthma[1]. A line of mice devoid of eosinophils was recently created to confirm hypotheses that a causative link exists between eosinophils and asthma-related pathogenesis[2]. Eotaxin is a potent eosinophil chemoattractant causing selective infiltration of these cells into the lungs. It binds to CC-chemokine receptor-3 (CCR3) that appears to be restricted to the eosinophil in mice[3]. Therefore, the eotaxin/CCR3 pathway is a potentially important target for asthma therapy.
We explored the effect of a CCR3 monoclonal antibody (CCR3 mAb) on airway eosinophilia and goblet cell hyperplasia (GCH)/mucus overproduction, the 2 most important hallmarks in asthma. The MUC5AC gene expression is thought to be a marker of GCH[4], and the epidermal growth factor receptor (EGFR) pathway is involved in mucin production in the airways[5]. In this study, we also examined the effect of CCR3 mAb on MUC5AC and EGFR gene expression in the lungs of asthmatic mice.
Materials and methods
Reagents Rat anti-mouse CCR3 mAb[6] was received as a gift from Dr James J LEE (Mayo Clinic, Phonex, Arizona, USA). Nonspecific rat IgG (ns-IgG) was purchased from Chemicon Co (Temecula, CA, USA); chicken ovalbumin (OVA, grade V) was from Sigma Chemicals (St Louis, MO, USA); inject alum was from Pierce Co (Rockford, IL, USA); IL-5 ELISA kit was from R&D Co (Minneapolis, MN, USA); Trizol for extracting RNA was from Invitrogen Co (Carlsbad, CA, USA).
OVA sensitization and challenge Male C57BL/6 mice, weighing 18–23 g, were purchased from the Shanghai Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China). The mice were sensitized and challenged with chicken OVA as previously described[7]. All the mice were sensitized by intraperitoneal injections (100 µL) of 80 µg OVA emulsified in 2 mg of Inject Alum on d 0 and d 14. The mice were exposed for 30 min to 1% OVA aerosol generated by an ultrasonic nebulizer on d 24–26. Blank control mice were sensitized and challenged with normal saline. In the preliminary experiment, we found that 6 µg/kg-mice of CCR3 mAb intraperitoneal administration resulted in over 90% reduction of eosinophils in the peripheral blood and bronchial alveolar lavage fluid (BALF). In order to strengthen the inhibition effect of CCR3 mAb on eosinophils, the mice were injected intraperitoneally with 150 µg of CCR3 mAb, then 50 µg of CCR3 mAb was mixed with the OVA solution and administered with each of the 3 OVA challenges in our formal experiment. Negative control mice were administered the same amounts of rat ns-IgG by both routes.
Cell count in the BALF Mice were sacrificed for sample collection on d 28. The tracheas were cannulated, the right lungs were ligated with a suture thread, and the left lobes of lungs were lavaged 3 times with 0.3 mL of ice-cold phosphate buffered saline (PBS). BALF 10 µL was used for the total cell counts and the residual BALF was centrifuged. The cell pellet was used to prepare cell slides by cytospin and stained with methylene blue/eosin. Four hundred cells were counted for estimating the differential counts of various cell types afterwards. The supernatant was preserved at -70 ºC for cytokine detection. The right lungs without lavage were stored at -70 ºC for total RNA isolation.
Lung histology The left lobes of the lungs were fixed in 10% buffered formalin. After embedded in paraffin, the tissues were cut into 4 µm thick sections. The lung sections were stained with hematoxylin/eosin (HE) and Alcian blue/periodic acid-Schiff (AB/PAS), respectively. Infiltrating lung eosinophils in parasagittal sections with HE staining were counted under microscopy. The goblet cell percentage (GCP) and airway mucus index (AMI) were measured as follows: GCP=the amount of goblet cells/total number of airway epithelial cells; AMI=the area of airway epithelium with AB/PAS staining/total area of the conducting airway epithelium. The analysis of the mucus content of the airway epithelium of mice from different groups was preformed using the imaging program of Leica Co (Wetzlar, Hessen, Germany).
Cytokine assay Interleukin (IL)-5 levels in the BALF were measured by ELISA method according to the manufac-turer’s protocol for users. The limit of IL-5 assay was 10 pg/mL.
Semi-quantitative RT-PCR The total RNA was extracted from about 0.2 mg of the lung tissues with Trizol reagents. For cDNA synthesis, 20 µL RT mixture containing total RNA 2 µg, dNTP 1 mmol/L, Olig(dt)18 prime 0.2 µg, RNasin 20 U, M-MLV reverse transcriptase 200 U was incubated at 42 ºC for 60 min, then the reverse transcriptase was inactivated by heating the reaction mixture at 72 ºC for 15 min. The expression of MUC5AC and EGFR mRNA in the lungs was measured by RT-PCR. Oligonucleotide primers specific for mouse MUC5AC, EGFR and glyceraldehyde phosphate dehydrogenase (GAPDH, an internal control) were synthesized according to published sequences: MUC5AC: sense: 5' CAG CCG AGA GGA GGG TTT GAT CT 3', anti-sense: 5' AGT CTC TCT CCG CTC CTC TCA AT 3'[4]; EGFR: sense: 5' GTG TGA AGA AGT GCC CCC GAA AC 3', anti-sense: 5' AAC GAC CGC CAA AGA AAA CTG ACC 3'[8]; GAPDH: sense: 5' CTG GTG CTG AGT ATG TCG TG 3', anti-sense: 5' CAG TCT TCT GAG TGG CAG TG 3'[4], with the product sizes 389 bp, 452 bp and 296 bp, respectively. PCR was performed as follows: 1 µL of cDNA mixture was subjected to amplification in 50µL of final volume with MgCl2 1.5 mmol/L, dNTPs 0.2 mmol/L, 20 pmol of each primer, and 2 U of Taq DNA polymerase in the reaction buffer on a PE2400 cycler (Perkin-Elmer, Foster City, CA, USA). The condition of PCR reactions was used below: 94 ºC for 5 min; then 94 ºC for 1 min; 56 ºC for MUC5AC, 58 ºC for EGFR and 60 ºC for GAPDH for 1min; 72 ºC for 45 s for 30 cycles (GAPDH) or 35 cycles (MUC5AC and EGFR) and 72 ºC for 10 min to end the reaction. PCR products were electrophoresed by 1.5% agarose gel containing ethidium bromide. The density of each band was measured by Gel-pro analyzer 4.0 (Exon-Intron, Inc., Loganville, PA, USA). The results were normalized as ratios compared with GAPDH.
Statistical analysis All data were presented as mean± SEM. One-way ANOVA was used for statistical analysis of the differences between the groups. A value of P<0.05 was considered statistically significant.
Results
Effect of CCR3 mAb on airway eosinophilic inflammation Numerous eosinophils appeared around the bronchioles and blood vessels in asthmatic mice whereas smooth airway walls and regular epithelium without inflammatory cell infiltration were seen in the saline control group. CCR3 mAb treatment obviously attenuated eosinophil infiltration, however, no significant difference was observed between the ns-IgG-treated mice and OVA-challenged mice (Figure 1). Eosinophils significantly increased and became the predominant inflammatory cells in the BALF and lung tissues of the OVA-challenged mice. CCR3 mAb treatment significantly reduced the number of total cells and eosinophils in the BALF and eosinophils number in the lung parenchyma (Table 1), but the administration of CCR3 mAb had no obvious effect on other cell types including lymphocytes, neutrophils and macrophages (data not shown).


Full table
Effect of CCR3 mAb on GCH and mucus overproduction The pathological results from the lung tissues showed that CCR3 mAb treatment significantly alleviated epithelial hypertrophy and mucus production in the airways of OVA-challenged mice compared with the ns-IgG treatment mice (Figure 2). However, GCP and AMI of the airway epithelium of CCR3 mAb-treated mice were still higher than those of the saline-challenged control mice (Table 2).


Full table
Effect of CCR3 mAb on IL-5 levels in the BALF The increased IL-5 levels were detected in the BALF of OVA-challenged mice vs the saline-treated mice. However, the IL-5 concentration in the BALF in CCR3 mAb and the ns-IgG-treated mice exhibited no obvious changes compared with the OVA-treated animals (Table 3).
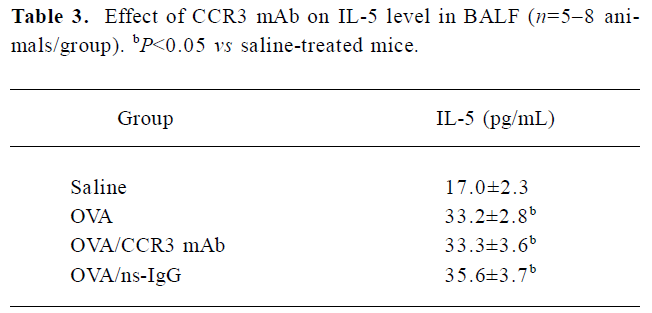
Full table
MUC5AC and EGFR mRNA expression in the lungs of mice The upregulated MUC5AC and EGFR mRNA expression was found in the OVA-challenged mice. CCR3 mAb treatment decreased the expression of MUC5AC and EGFR mRNA. No obvious difference of MUC5AC and EGFR mRNA expression was observed in the ns-IgG-treated mice compared with the OVA-challenged mice (Figures 3, 4).


Discussion
Although the key role of eosinophil in allergic pulmonary diseases is challenged by the clinical trials of the anti-IL-5 monoclonal antibody[9], more and more data from both asthma patients and mouse models have demonstrated that airway eosinophilia infiltration is one of the most important characteristics of asthma. Human eosinophils mediate pathogenic effects from several independent mechanisms that allow the eosinophil to modulate the intensity of pulmonary inflammation, as well as elicit cell death and loss of structural integrity of the lung, leading to pulmonary dysfunction[10]. Recently, a line of mice devoid of eosinophils was created to confirm hypotheses that a causative link exists between eosinophils and asthma-related pathogenesis such as airway epithelial pathology, mucus hypersecretion and AHR in Lee Laboratory (Mayo Clinic, Phonex, Arizona, USA)[2,7]. Therefore, blocking the onset and progression of asthma by specifically depleting eosinophils is effective in asthma therapy.
The chemokine receptor CCR3 seems to play a major role in allergic diseases which is supported by the detection of this receptor on human eosinophils, Th2 cells, mast cells and basophils[11–13]. However, Grimaldi et al revealed that mouse CCR3 was expressed on eosinophils, but not on stem cells, dendritic cells or cells from the thymus, lymph node, or spleen of normal mice by flow cytometry. Unlike human Th2 cells, mouse Th2 cells did not express detectable levels of CCR3 nor did they give a measurable response to eotaxin[3]. The reactivity of CCR3 mAb in our experiment was proven to be restricted to eosinophils[3,6].
Our study showed that dual administration (intraperitoneal injection and aerosol inhalation) of CCR3 mAb can effectively ablate eosinophils in the BALF and significantly reduce lung eosinophilia relative to OVA-challenged mice. In addition, CCR3 mAb was found to have no observable effect on the number of other cell types (lymphocytes, macrophages and neutrophils) in the BALF and lung tissues, indicating the specificity of mouse CCR3 mAb treatment for only eosinophils. Using the CCR3-deficient mouse model, Humbles et al found that CCR3 depletion significantly abrogated eosinophil recruitment to the lung after allergen challenge, with the majority of the eosinophils being arrested in the subendothelial space[14]. Similarly, a CCR3 monoclonal antibody and small molecule CCR3 antagonists were demonstrated to be capable of reducing pulmonary eosinophilic inflammation in mouse allergenic models of airway inflammation[15]. Taken together, CCR3 antagonism has the potential to influence eosinophil infiltration into the lungs of asthmatics and is therefore a promising therapeutic target.
Shi and colleagues pointed out that eosinophils were able to express major histocompatibility complex II complex and costimulatory molecules and function as antigen-presenting cells (APC)[16]. Eosinophils have the potential to activate lung Th2 cells to release disease-modulating cytokines[17], but the investigation from van Rijt et al did not support any role for airway eosinophils as APC to naive T cells, despite their migration to the draining lymph nodes at times of allergen exposure[18]. In our study, Th2 cytokine (IL-5) levels in the BALF from CCR3 mAb-treated animals were unaffected compared with the OVA-treated mice. Justice et al also proved that the anti-CCR3-mediated loss of eosinophils from the lung did not have a demonstrable effect on the activities of T and B lymphocytes[6]. So we think that eosinophils function as APC in the modulation of T cell activities which may be relatively limited and can be compensated by other APC (dendritic cells, macrophages and B lymphocytes) in the absence of eosinophils because dendritic cells are clearly superior in activating T cells in the draining lymph nodes of the lung[18].
Globlet cell metaplasia (GCM), GCH and mucus hypersecretion are prominent features of allergic asthma[19]. Numerous inflammatory mediators produced in asthmatic airways, together with neural mechanisms, can theoretically increase mucin secretion, induce plasma exudation, upregulate MUC gene expression (MUC5AC gene expression is thought to be a marker of GCH[5]), increase mucin synthesis and cause GCH[20,21]. Most of the above pathways operate through the EGFR and its tyrosine kinase intracellular signaling cascade[22]. EGFR and MUC5AC expression at both gene and protein levels is upregulated in goblet cells in bronchial biopsies from asthmatic patients compared with non-asthmatic persons[23]. Burgel et al demonstrated that activated human eosinophils induced mucin synthesis in human airway epithelial cells via EGFR activation[24]. We analyzed MUC5AC and EGFR gene expression in the lungs of asthmatic mice. Over-expression of MUC5AC and EGFR mRNA in the lungs was found in the OVA-treated mice compared with the saline-treated animals, and CCR3 mAb treatment significantly reduced MUC5AC and EGFR mRNA levels in the lungs of OVA-challenged mice by the ablation of eosinophils relative to ns-IgG-treated mice. Therefore, we deduce that CCR3 mAb may downregulate MUC5AC gene expression, at least partly, through the EGFR pathway.
Our results show that CCR3 mAb-mediated ablation of eosinophils in OVA-treated mice can significantly reduce GCM/GCH and mucus overproduction compared with ns-IgG-treated mice, indicating that the products of eosinophils are capable of stimulating an increase in goblet-cell number and mucus secretion. However, the GCP and AMI of CCR3 mAb-treated mice were still higher than those of the normal saline-treated mice, suggesting the existence of both eosinophil-independent and eosinophil-dependent pathways leading to allergen-mediated goblet cell metaplasia/mucus production[6].
In summary, CCR3 mAb can effectively ablate lung eosinophilia and significantly inhibit mucus overproduction in a mouse model of asthma. Interfering with the CCR3 receptor may represent a potential new therapy for the treatment of asthma.
References
- Lungarella G, Menegazzi R, Gardi C, Spessotto P, de Santi MM, Bertoncin P, et al. Identification of elastase in human eosinophils: immunolocalization, isolation, and partial characterization. Arch Biochem Biophys 1992;292:128-35.
- Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O’Neill KR, et al. Defining a link with asthmain mice congenitally deficient in eosinophils. Science 2004;305:1726-9.
- Grimaldi JC, Yu NX, Grunig G, Seymour BW, Cottrez F, Robinson DS, et al. Depletion of eosinophils in mice through the use of antibodies specific for C-C chemokine receptor 3 (CCR3). J Leukoc Biol 1999;65:846-53.
- Kibe A, Inoue H, Fukuyama S, Machida K, Matsumoto K, Koto H, et al. Differential regulation by glucocorticoid of interleukin-13-induced eosinophilia, hyperresponsiveness, and goblet cell hyperplasia in mouse airways. Am J Respir Crit Care Med 2003;167:50-6.
- Takeyama K, Dabbagh K, Lee HM, Agusti C, Lausier JA, Ueki IF, et al. Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci USA 1999;96:3081-6.
- Justice JP, Borchers MT, Crosby JR, Hines EM, Shen HH, Ochkur SI, et al. Ablation of eosinophils leads to a reduction of allergen-induced pulmonary pathology. Am J Physiol Lung Cell Mol Physiol 2003;284:169-78.
- Shen HH, Ochkur SI, McGarry MP, Crosby JR, Hines EM, Borchers MT, et al. A causative relationship exists between eosinophils and the development of allergic pulmonary pathologies in the mouse. J Immunol 2003;170:3296-305.
- Honda J, Oomizu S, Kiuchi Y, Komatsu N, Takeuchi S, Takahashi S. Identification of epidermal growth factor mRNA-expressing cells in the mouse anterior pituitary. Neuroendocrinology 2000;71:155-62.
- Leckie MJ. Anti-interleukin-5 monoclonal antibodies: preclinical and clinical evidence in asthma models. Am J Respir Med 2003;2:245-59.
- Lee NA, Gelfand EW, Lee JJ. Pulmonary T cells and eosinophils: Coconspirators or independent triggers of allergic respiratory pathology? J Allergy Clin Immunol 2001;107:945-7.
- Elsner J, Escher SE, Forssmann U. Chemokine receptor antagonists: a novel therapeutic approach in allergic diseases. Allergy 2004;59:1243-58.
- Uguccioni M, Mackay CR, Ochensberger B, Loetscher P, Rhis S, LaRosa GJ, et al. High expression of the chemokine receptor CCR3 in human blood basophils. Role in activation by eotaxin, MCP-4, and other chemokines. J Clin Invest 1997;100:1137-43.
- Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science 1997;277:2005-200.
- Humbles AA, Lu B, Friends DS, Okinaga S, Lora J, Al-Garawi A, et al. The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness. Proc Natl Acad Sci USA 2002;99:1479-84.
- Das AM, Vaddi KG, Solomon KA, Krauthauser C, Jiang X, McIntyre KW, et al. Selective inhibition of eosinophil influx into the lung by small molecule CCR3 antagonists in mouse models of allergic inflammation. J Pharmacol Exp Ther 2006;318:411-7.
- Shi HZ. Eosinophils function as antigen-presenting cells. J Leukoc Biol 2004;76:520-7.
- Xie ZF, Shi HZ, Qin XJ, Kang LF, Huang CP, Chen YQ. Effects of antigen presentation of eosinophils on lung Th1/Th2 imbalance. Chin Med J 2005;118:6-11.
- van Rijt LS, Vos N, Hijdra D, de Vries VC, Hoogsteden HC, Lambrecht BN. Airway eosinophils accumulate in the mediastinal lymph nodes but lack antigen-presenting potential for naive T cells. J Immunol 2003;171:3372-8.
- Rogers DF. Airway hypersecretion in allergic rhinitis and asthma: new pharmacotherapy. Curr Allergy Asthma Rep 2003;3:238-48.
- Jackson AD. Airway goblet-cell mucus secretion. Trends Pharmacol Sci 2001;22:39-45.
- Rogers DF. Pharmacological regulation of the neuronal control of airway mucus secretion. Curr Opin Pharmacol 2002;2:249-55.
- Nadel JA, Burgel PR. The role of epidermal growth factor in mucus production. Curr Opin Pharmacol 2001;1:254-8.
- Takeyama K, Fahy JV, Nadel JA. Relationship of epidermal growth factor receptors to goblet cell production in human bronchi. Am J Respir Crit Care Med 2001;163:511-6.
- Burgel PR, Lazarus SC, Tam DC, Ueki IF, Atabai K, Birch M, et al. Human eosinophils induce mucin production in airway epithelial cells via epidermal growth factor receptor activation. J Immunol 2001;167:5948-54.