
Construction of a small peptide library related to inhibitor OM99-2 and its structure-activity relationship to β-secretase1
Introduction
With increasing life expectancy, Alzheimer’s disease (AD) will become a heavy burden to families and society. AD is a neurodegenerative disorder clinically characterized by progressive dementia that inevitably leads to incapacitation and death. Until now, therapies have temporarily improved cognitive function, but have done little to block the progression of AD[1]. Understanding the pathophysiological mechanisms that underlie neurodegeneration in AD is essential for the rational design of therapies to retard or halt progression of the disease. The accumulation of β-amyloid (Aβ) in the human brain is considered to be the main cause of the development of AD[2]. The β-amyloid (40–42 residues) is generated in vivo through the proteolytic cleavage of the membrane-anchored β-amyloid precursor protein (APP) by β- and γ-secretase[3]. Both proteases are potential targets for inhibitor drugs against AD, but β-secretase, which cleaves APP on the luminal side and is considered to be the rate-limiting step in the processing of APP to Aβ peptide[4,5], is considered to be the major therapeutic target. β-secretase and the crystal structure of its catalytic domain were also determined[3]. Three transition–state analog inhibitors of β-secretase have been reported, including: (1) statine structure-related compounds[6-8]; (2) hydroxyethylene (HE) structure-related compounds[9,10]; (3) hydroxyethylamine structure-related compounds[11].
OM99-2 is the first inhibitor incorporating 8-residue (Molecular weight: about 900 Da) and a non-hydrolyzable Leu-Ala HE dipeptide isostere as depicted in Figure 1. This compound has an IC50 of 80 nmol/L for β-secretase and is a product designed on the basis of the common enzymatic mechanism of aspartic proteases. Substrate transition-state mimics have been widely used for the design of aspartic protease inhibitors, but enzyme inhibitors with therapeutic potential are preferable as they have low molecular weight and are available compounds for oral administration. Therefore, the inhibitor-like OM99-2 is not viable drug candidate, although it is a good lead compound for rational drug design efforts.
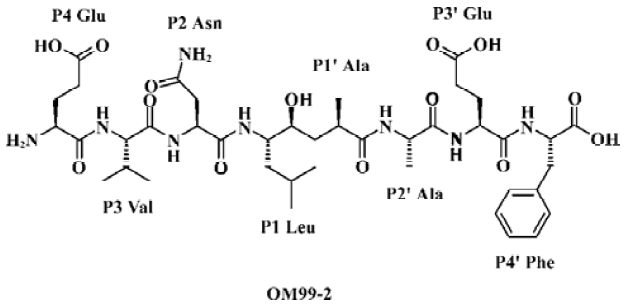
In the current design, we planned to synthesize a combinatorial inhibitors library having residues from 2 to 8 (Figure 2). Combinatorial chemistry techniques were utilized to build this 16-compound (4×4) library, keeping “P4-P3-P2-P1'”, “P3-P2-P1'”, “P2-P1'”, “P1'” on the left side and “P1'”, “P1'-P2'”, “P1'-P2'-P3'”, “P1'-P2'- P3'-P4'” on the right side, respectively. When the P' side only contains P1', the inhibitors are easily transferred from γ-hydroxy acid to the corresponding lactone. We therefore turned it into methylamide with a methylamine nucleophilic attack on the lactone. Compounds with various lengths on both sides were prepared in order to check the biological effect.
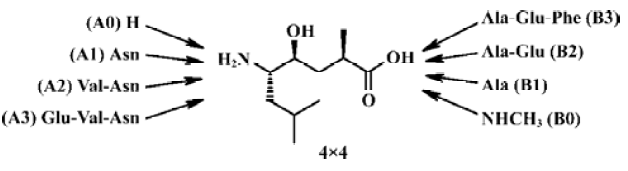
In the present study, we systematically launched structure-activity relationship (SAR) investigations surrounding 8 subsites. In order to further investigate the geometrical and energetical characteristics of the binding between β-secretase and OM99-2, the computational methods, molecular dynamics (MD) and molecular mechanics-generalized born surface area (MM-GBSA) were used. This method was used extensively to provide the binding information in many biological studies on protein-protein interactions, protein-DNA interactions and receptor-drug interactions[12–14]. Based on the crystal structure of the complex[3], we utilized this method to compute the molecular dynamics trajectory and post-processed it with MM-GBSA. The results calculated from this simulation were consistent with the results from the combinatorial peptide library. This enables us to study in detail the interactions between β-secretase and OM99-2, and further assist peptide modification towards the oral drug with low molecular weight required for AD treatment.
Materials and methods
Chemistry The HE dipeptide isostere (4, Figure 3) was produced from commercially available L-leucine, and was synthesized by the methods reported previously[15]. The acid was transferred to the corresponding active ester to improve reactivity. All library compounds were prepared using Wang resin, a general synthetic route employed for the preparation as shown in the Figure 4. The linkage of the B group was conducted under general coupling conditions and the solid resin containing B1, B2, B3 reacted with 5 to afford the intermediate 6. After that, A1, A2, A3 were introduced to 6, respectively. The removal of the N-Fmoc group was carried out in 20% piperidine in N,N-dimethylforamide (DMF) for 15 min; the removal of the silyl protective group was as a result of treatment with tetrabutylammonium fluoride in tetrahydrofuran (THF) to give 7. The Kaiser reagent[16] test was used to monitor the reaction to ensure peptide homogeneity. The tert-butyl ester and trityl were removed while the final products were cleaved from the Wang resin with 50% trifluoroacetic acid in CH2Cl2. The products were isolated via filtration from the resin and the filtrate was added to 5 mL ethyl ether; 9 was treated with methylamine (solution in methanol) overnight at room temperature (Figure 4). The resulting crude products were collected and purified via semipreparative reverse phase High Performance Liquid Chromatography (HPLC). All compounds were evaluated by a β-secretase enzyme assay.
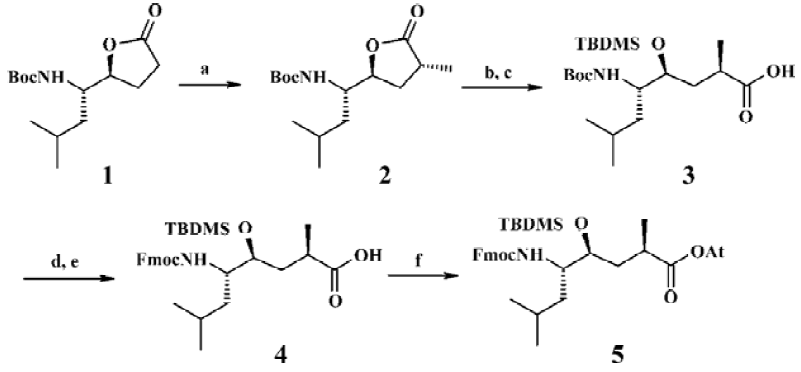
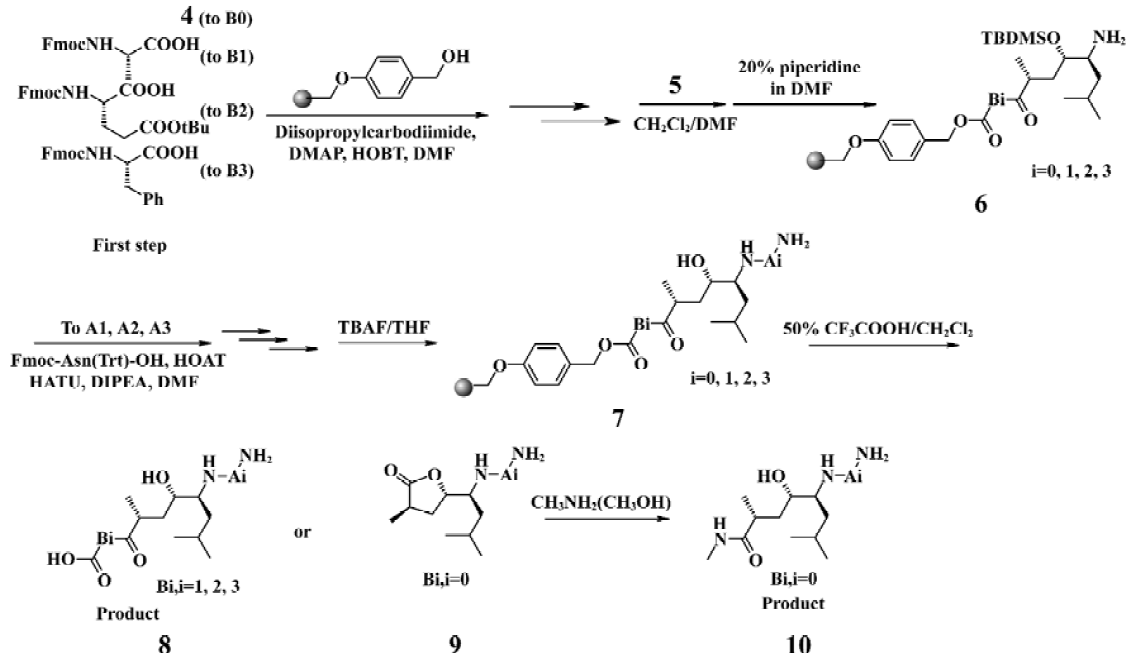
Solid-phase synthesis protocol was utilized for the synthesis of all inhibitors. The synthesis was initiated with Wang resin (1.2 mmol/g). The coupled residues included N-Fmoc-L-Phe, N-Fmoc-L-Glu (t-butyl ester), N-Fmoc-L-Ala, the HE segment prepared by ourselves, N-Fmoc-L-Asn (trityl), N-Fmoc-L-Val and N-Fmoc-L-Glu (t-butyl ester).
Biological assays Recombinant human β-secretase ectodomain (amino acid residues 1–460) was expressed as a secreted protein with a C-terminal His tag in insect cells using baculovirus infection. With sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the apparent molecular masses were approximately 53 kDa after purification. The FRET substrate H-Arg-Glu (EADANS) -Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys(DABCYL)-Arg-OH for β-secretase was purchased from Calbiochem Co (San Diego, California, USA). The standard assay volume was 100 µL including 100 mmol/L sodium acetate, pH 4.0, 50 nmol/L β-secretase, 5 µmol/L substrate and different inhibitor concentrations. The reaction was initiated by the addition of the substrate and fluorescence intensity was measured using a Wallac Victor plate reader (Perkin-Elmer, Wellesley, Massachusetts, USA) at Ex355/Em460 after 60 min incubation at 25 °C. IC50 was calculated from the non-linear curve fitting of percentage inhibition vs inhibitor concentration [I] by using the following equation:
% inhibition=100/{1+(IC50/[I])k}, where k is the Hill coefficient (1)
Computational simulation The crystal structure of the complex of β-secretase and OM99-2 (PDB access code: 1FKN) was retrieved from the Protein Data Bank[3]. The abnormal residues in the peptide analog OM99-2 were extracted from the structure and treated with Gaussian98 to optimize at the 6–31G level. The atomic partial charges were fitted to the electrostatic potential from the Gaussian calculation with the RESP method. The Amber parameter files for HE were generated with the program antechamber which combined the Amber general force field and the RESP charge[17]. To determine the protonation state of the titratable residues, especially the active site Asp32, Asp228, the program MEAD was adopted to calculate the pKa value of these residues. According to the result, Asp32 should be in a protonated state. The missing hydrogens of the other residues were added according to the Amber residue template. The 6 cysteine residues formed 3 disulfide bonds in the crystal structure which were modeled properly as CYX residues in the Amber force field. The program XLeap was used to prepare the Amber topology and coordinate files. The complex was then solvated with a TIP3P water box which extended at least 10 Å away from the any atoms in the solute complex. Finally, 10 sodium ions were added to neutralize the whole solvated system. This resulted in a system of about 86 Å×90 Å×82 Å, which contained approximately 15 000 water molecules and protein-peptide analog complex.
The Sander program in the Amber software package was used to gradually minimize the whole system with the steepest descent method followed by the conjugate gradient. First, the enzyme-peptide analog complex was fixed and only water molecules were allowed to optimize to release the conflict possibly exited between the solute and the water molecules. The complex then gradually reduced the constrained force in 20 000 steps followed by 30 000 steps of relaxation of the whole system without any constraint. The relaxed structures were then subjected to MD simulations. The system was gradually heated from 0 K to 300 K in 100 ps, and then equilibrated for 200 ps at 300 K, followed by a 1500 ps data collection run, giving a total simulation time of 1800 ps. The non-bonded cutoff was set to 8.0 Å, and the non-bonded pairs were updated every 25 steps. The SHAKE method was applied to constrain all covalent bonds involving hydrogen atoms[18]. The simulation was coupled to a 300 K thermal bath at 1.0 atm pressure by applying the algorithm of Berendsen et al[19]. The temperature and pressure coupled parameters were set as 0.2 ps and 0.05 ps, respectively. An integration time step of the MD calculations was 2 fs. In the energy minimizations and MD simulations, periodic boundary conditions were applied for all directions.
The computed trajectory of the production phase was post-processed to calculate the binding free energy and decomposition of the free energy to every residue in the complex by using the MM-GBSA method implemented in Amber 7.0 according to the following equation:
ΔG=Gcomplex-(Genzyme+Ganalog)
G=Ggas+Gsolvation-TS
Ggas≈Etotal=Eband+Eangle+Etor+EVDW+Eele (2)
Gsolvation=GGB+Gnon-polar
Gnon-polar=γ×SA+b
where Gcomplex, Genzyme, Ganalog are the free energy of the complex, enzyme and analog, respectively. Every absolute free energy is calculated from the free energy in the gas state Ggas and salvation free energy Gsolvation and entropy term TS. Ggas is associated with the Amber force field energy, while Gsolvation consists of polar and non-polar contributions. The polar term is calculated with the generalized Born model, and the non-polar term is calculated from the empirical equation related to the molecular surface area of the system. The γ is set to 0.00542 kcal·mol-1·Å-2 and the b is set to 0.092 kcal/mol. The entropy of the system is calculated according to the standard statistics mechanics method described in every statistics mechanics textbook.
Due to the limitation of the computational resource, every 15 ps snapshot was extracted from the product trajectory, which resulted in a total of 100 conformations subjected to the MM-GBSA analysis. The entropy calculation was extremely time-consuming for the large system, so the subsystem contained peptide analog and the residues 8 Å close to the analog. Only 4 of the 100 conformations (1, 500, 1000, and 1500 ps) were used to build the subsystem and the program NMODE was used to minimize the subsystem and calculate the entropy.
Results and Discussions
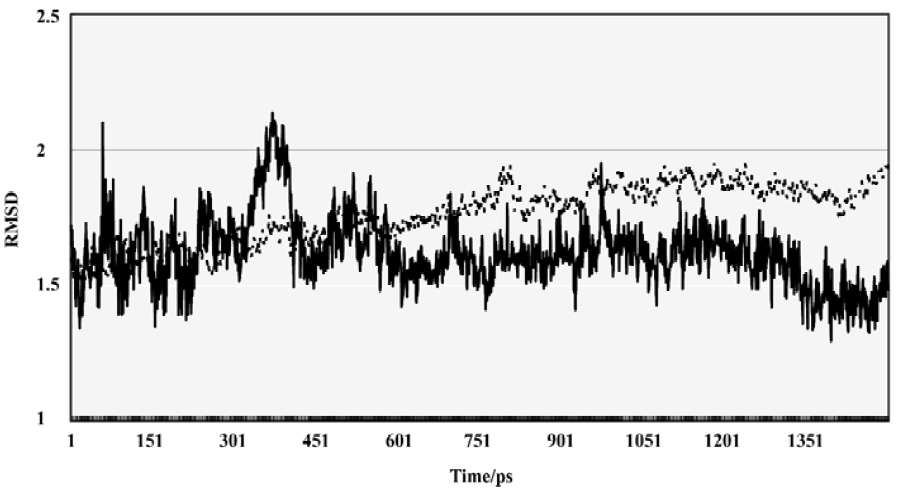
Conformational and energetic features of the enzyme-substrate complex The complex of β-secretase and OM99-2 was performed with MD simulation in order to evaluate the conformational flexibility. The overall mass weight root mean square deviation (RMSD) was used to assess the stability of the whole system. As shown in Figure 5, after the first 500 ps equilibrization phase, the evolution of the RMSD over the simulation time clearly showed the system reaching a stable state, and the overall RMSD, with respect to the original crystal structure, was about 1.8 Å. The RMSD of the peptide analog also demonstrated that the ligand was in a stable conformational state after the first 500 ps. From the quantity of RMSD data, it was found that the peptide analog showed less deviation from the crystal structure.
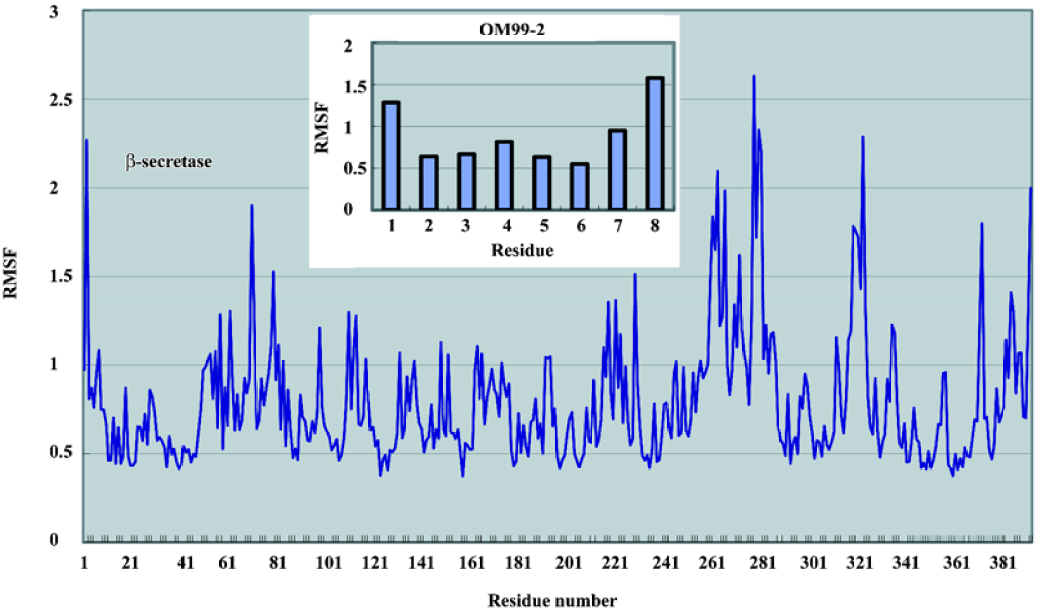
The root mean square fluctuation (RMSF) of the residues over the last 1000 ps was also calculated to determine the flexible parts in the complex structure. As depicted in Figure 6, the N-lobe of the catalytic domain of the β-secretase was more rigid than the C-lobe. The most obvious flexible parts in the N-lobe were the two β strands and the connecting loop which covered the binding site. As many previous studies have shown, this loop is important to the substrate entry and binding. Disclosed by several crystal structures, this loop can adopt an induced-fit effect to lift and accommodate a slightly large substrate or inhibitor in the binding site. In the C-lobe, there are 2 significant segments; residues 254–275 and 312-318 which show large fluctuations compared with the overall structure. In the 3D structure, these parts are located on the surface of the β-secretase which may only reflect the intrinsic flexibility of these loop structures. The fluctuation of the ligand clearly demonstrated a pattern that the further from the middle P1 and P1' are, the greater the fluctuation. However, from a quantitative point of view, the fluctuation of these residues is much smaller than the flexible parts in the β-secretase. From these comparisons, it was indicated that the deviations of the OM99-2 terminals were still within a reasonable range during the simulation time. The overall conformational transitions from 1 part indicated that simulations from 500 ps to 1500 ps were stable, and from another part, the dynamic features of the complex structure were shown, which may further assist the drug design process.
Protein-ligand interactions To investigate the protein-ligand interaction, the interface between the β-secretase and OM99-2 was analyzed to compute the surface area change over the simulation time. A total of 100 snapshots were extracted from 500 ps to 1500 ps and calculated with the MSMS program. The average complex surface area was about 17022.8 Å, while those of β-secretase and OM99-2 are about 17383.5 Å, 1296.7 Å, respectively.
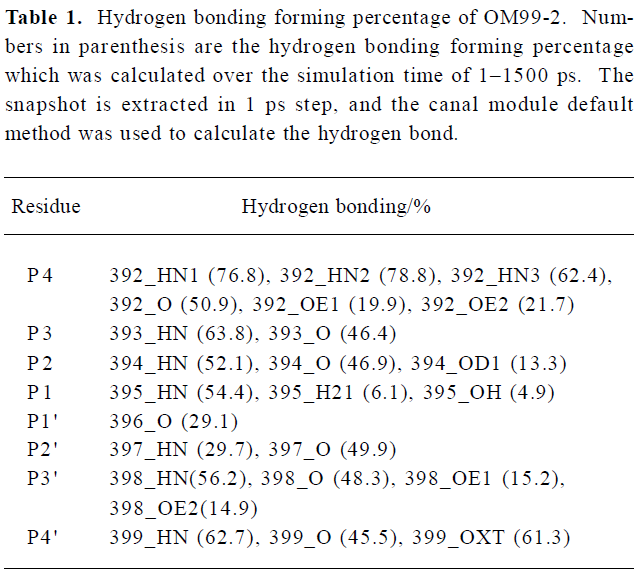
To investigate the interaction between β-secretase and OM99-2 in detail, the hydrogen bonding interactions were clustered based on every residue in OM99-2. The results are present in Table 1, which shows the subsites P4, P2, P3' having more hydrogen bonding interaction with the enzyme, while the other subsites, especially P1' and P2' demonstrated their hydrophobic characteristics. The P4 subsite makes many hydrogen bonding interactions with the residues of β-secretase such as Thr232 and Arg307, which is consistent with findings that P4 glutamic acid has a large contribution to the binding free energy as described in the GBSA analysis section below. The other important contributions come from P2 and P3'; among these interactions, the flap structure has lots of interactions with OM99-2, including residues 71–73. This reflects the importance of the flap and may be explored in designing the specific ligand to the β-secretase. Even the catalytic residues Asp32 and Asp228 are in subsites S1 and S1'; but only make hydrogen bonding with the hydroxyl of P1. The interactions involving subsites S1 and S1' are VDW interactions which come from Leu30, Phe108, Gly34, and the tip of the flap. These detailed interactions in the binding site would be valuable for further ligand design.
Full table
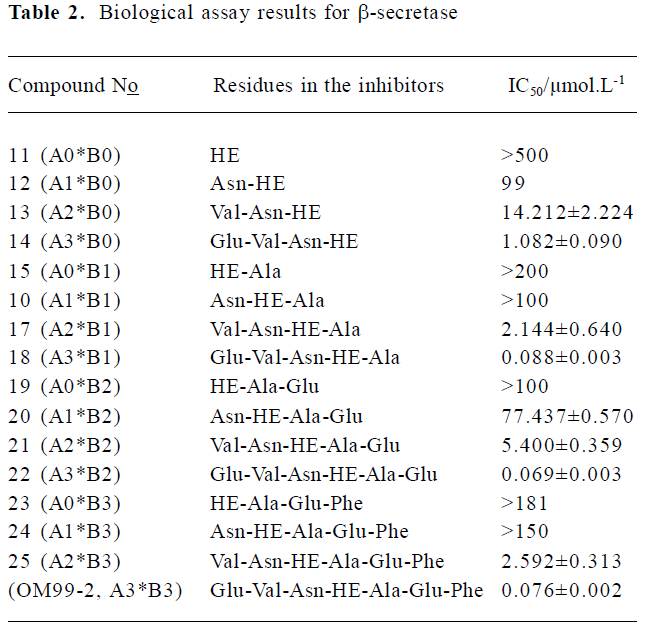
GBSA analysis and biological assay The biological assay results about the N-terminal and C-terminal SAR of the library inhibitors are summarized in Table 2 (11-OM99-2). The shortest compound 11, which includes only the P1-P1' residues, gives an almost undetectable -secretase inhibitory activity (IC50>500 µmol/L). After adding P2 group to 12 (IC50=99 µmol/L), the activity increased more than 5-fold. Further increasing the length of the left side to P3, P4 (compounds 13, 14) led to 7-fold and 13-fold activity increase, respectively (IC50=14.2 µmol/L and 1.08 µmol/L). As shown in Table 1, when compared to 20, 21, 22 (A3*B2), the addition of P3 and P4 resulted in 14-fold potency enhancement and had the most dramatic effect with a 78-fold improvement between 21, 22 (IC50=77.4 µmol/L→5.4 µmol/L→0.069 µmol/L); the same rule was observed in 24, 25, OM99-2.
Full table
However, with the right side subsites SAR, we studied the preference of the P' position about its ability for interaction with the aspartic protease. The inhibitor OM99-2 has high potency (control, IC50=0.076 µmol/L); thus, we decreased the residues of carboxyl-terminal gradually. First, we removed the P4' yielded inhibitor 22 (A3*B2) which has high potency (IC50=0.069 µmol/L). Further trunction of P3' gave inhibitor 18 (A3*B1; IC50=0.088 µmol/L) equal activity, but a 280 Da loss in molecular weight to OM99-2. Removal of the last right residue resulted in inhibitor 14 (A3*B0) yielding a moderate 1.08 µmol/L BACE enzyme inhibitory activity, implying that the right side residues play a minor role in the inhibitor-enzyme interaction. The same conclusion could be deduced from the compound assembly 25→21→17→13.
To analyze the energetical character of the binding, the MM-GBSA method was performed to 100 snapshots extracted from the trajectory over the last 1500 ps. The average contribution of each term is listed in Table 3. From an energy point of view, the gas interaction, which including bonded electrostatic VDW interactions, is a dominant factor in the overall binding free energy, while the solvation free energy and the entropy had negative contributions. These results suggest that if one can design a more rigid ligand, it might reduce the entropy lost and improve the affinity.

Full table
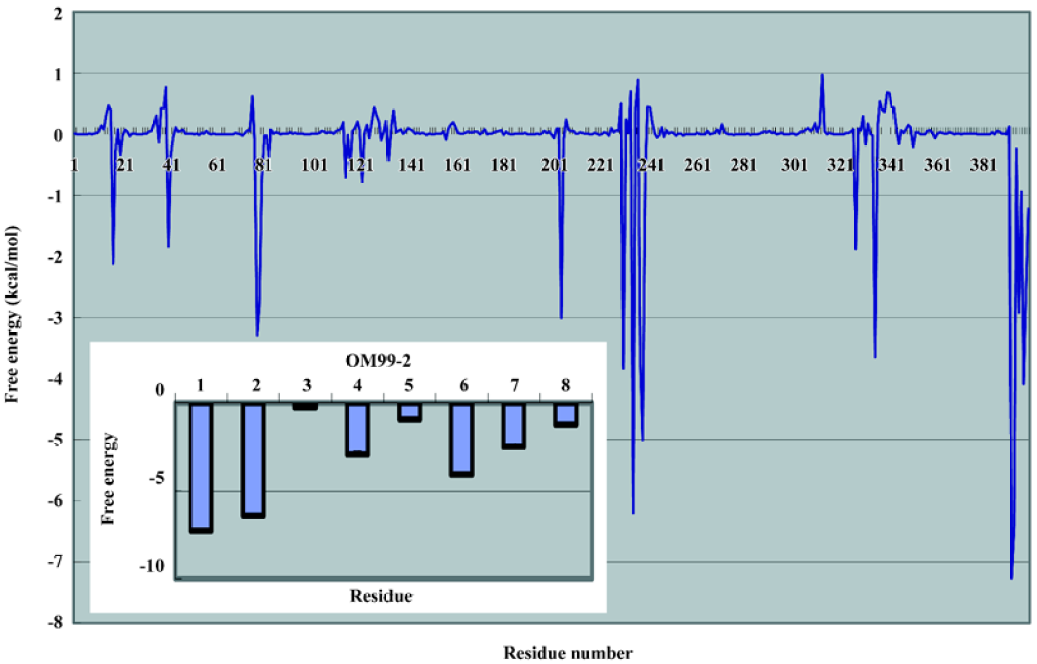
In order to examine the residue contribution to the whole binding, the free energy decomposition method implemented in the MM-PBSA module of the Amber package was used. The results are illustrated in Figure 7. The largest contribution in β-secretase are residues Asp228, Thr231, and Thr232. They are mostly due to the interaction with P1 and P1' through VDW interaction. The other important residues are Gly34, Tyr71, Thr72, GLn73, and Tyr198. These identified residues are consistent with the structural analysis. The free energy contributions of OM99-2 are also analyzed. We found that the P side residues were more significant than the P' side, among which the P4, P3, and P2' residues played an important role in the overall binding; the contributions were -7.27, -6.43, and -4.09 kJ/mol, respectively. Compared the biological data with the computational analysis, some important features were found. The P4 subsite revealed that the hydrophilic property and the terminus N atom formed several hydrogen bonds with β-secretase, making the P4 subsite crucial in the whole ligand. The hydrophobic residue P3 and P2' play an important role in overall binding, which matches the data set from the biological assay. It shows the P3 subsite almost as significant as the P4 subsite, and adding these residues improves the affinity by about 50%. The P1 and P1' residues do not contribute much to the binding free energy. This is complemented by the residues in β-secretase, such as 2 catalytic aspartate acid and the tip of the flap structure. Other residues such as P4' and P3' are not notable in the binding, and as suggested by other studies, these can truncate without much loss of affinity.
A substrate-based combinatorial library of β-secretase inhibitors were designed and synthesized taking the HE isostere as dipeptide isostere. The biological evaluation led to the discovery of fewer shorter potent inhibitors (18, 22 and 26). Computational methods, MD and MM-GBSA analysis, were also performed to assist in the understanding of the detailed binding. Importantly, the assay and computational results provide us with systematic data about binding specificity on the P side and the P' side, and show that P4 and P3 are vital to the binding as they have a large enough space to modify and achieve specificity and stronger affinity.
Acknowledgement
We thank Prof Hua-liang JIANG at the Shanghai Institute of Materia Medica for providing the computational facility to perform the molecular dynamics simulation.
References
- VanDenBerg CM, Kazmi Y, Jann MW. Cholinesterase inhibitors for the treatment of Alzheimer’s disease in the elderly. Drugs Aging 2000;16:123-38.
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 1999;399A:23.
- Hong L, Koelsch G, Lin XL, Wu SL, Terzyan S, Ghosh AK, et al. Structure of the protease domain of memapsin 2 (β-secretase) complexed with inhibitor. Science 2000;290:150-3.
- Selkoe DJ. Alzheimer’s disease: genes, proteins and therapy. Physiol Rev 2001;81:741-66.
- Vassar R, Citron M. Aβ-generating enzymes: recent advances in β- and β-secretase research. Neuron 2000;27:419-22.
- Hu JD, Cwi CL, Smiley DL, Timm D, Erickson JA, McGee JE, et al. Design and synthesis of statine-containing BACE-inhibitors. Bioorg Med Chem Lett 2003;13:4335-9.
- Tung JS, Davis DL, Anderson JP, Walker DE, Mamo S, Jewett N, et al. Design of substrate-based inhibitors of human β-secretase. J Med Chem 2002;45:259-62.
- Vmezawa H, Agoyagi T, Morishima H, Matzusaki M, Hamada H, Takeuchi T. Pepstatin, a new pepsin inhibitor produced by actinomycetes. J Antibiot 1970;23:259-62.
- Hom RK, Gailunas AF, Mamo S, Fang LY, Tung JS, Walker DE, et al. Design and synthesis of hydroxyethylene-based peptidomi-metic inhibitors of human β-secretase. J Med Chem 2004;47:158-64.
- Ghosh AK, Bilcer G, Harwood C, Kawahama R, Shin D, Hussain KA, et al. Structure-based design: potent inhibitors of human brain memapsin 2 (β-secretase). J Med Chem 2001;44:2865-8.
- Tamamura H, Kato T, Otaka A, Fujii N. Synthesis of potent β-secretase inhibitors containing a hydroxyethylamine dipeptide isostere and their structure-activity relationship studies. Org Biomol Chem 2003;1:2468-73.
- Wang W, Donini O, Reyes CM, Kollman PA. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Biophys Biomol Struct 2001;30:211-43.
- Zeng J, Treutlein HR, Simonson T. Molecular dynamics simulations of the Ras:Raf and Rap:Raf complexes. Proteins 1999;35:89-100.
- Reyes CM, Kollman PA. Structure and thermodynamics of RNA-protein binding: using molecular dynamics and free energy analyses to calculate the free energies of binding and conformational change. J Mol Biol 2000;297:1145-58.
- Ghosh AK, Shin D, Downs D, Koelsch G, Lin XL, Ermolieff J, et al. Design of potent inhibitors for human brain memapsin 2 (β-secretase). J Am Chem Soc 2000;122:3522-3.
- Atherton E, Sheppard RC. Solid phase peptide synthesis. Oxford, UK: Oxford University Press; p108.
- Case DA, Pearlman DA, Caldwell JW, Cheatham TEI, Wang J, Ross WS, . AMBER 7. San Francisco: University of California; 2002.
- Ryckaert JP, Ciccotti G, Berendsen JC. Numerical integration of the Cartesian equation of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1977;23:327-41.
- Berendsen HCJ, Postma JPM, van Gunsteren WF, DiNole A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984;81:3684-90.