
Granulocyte-macrophage colony-stimulating factor DNA prime-protein boost strategy to enhance efficacy of a recombinant pertussis DNA vaccine1
Introduction
Pertussis or whooping cough is a highly contagious disease of the human respiratory tract. There are approximately 45 million cases of whooping cough worldwide per year, of these 400 000 result in fatalities. Most cases of severe symptoms and the majority of fatalities are observed in early infancy[1]. Pertussis causes significant economic costs for families, hospitals and communities[2].
Whole-cell and acellular pertussis vaccines have been effective in providing protection against Bordetella pertussis infection in humans[3,4]. However, whole-cell vaccines have been associated with serious side effects such as convulsions and encephalopathy[5]. As a result, acellular vaccines have replaced whole-cell vaccines in many developed countries. Unfortunately, acellular vaccines are too expensive for routine use, especially in developing countries which have the greatest need. In the last 10 years, there has been a resurgence of pertussis in many developing countries.
B pertussis secretes several virulence factors, including toxins such as pertussis toxin (PT), tracheal cytotoxin and adhesins such as filamentous hemagglutinin (FHA) and pertactin (PRN)[6]. Protection for pertussis has been associated with higher levels of antibodies against PT, PRN and FHA[4]. PT is a protein exotoxin which is central to the pathogenesis of whooping cough. Detoxified PT is considered to be essential for acellular pertussis vaccines[3]. It is the S1 subunit of PT which possesses enzymatic capabilities of the ADP ribosyltransferase. A mutant containing a single amino acid substitution Arg9-Lys of pertussis toxin subunit 1 (PTS1) reduced the enzymatic activity of wild PTS1 by approximately 0.02%, while retaining the protective epitope[7]. The Arg9-Lys and Glu129-Gly analog was found to be significantly more immunogenic than other mutation analogs[8]. PRN is another important pertussis vaccine antigen in multicomponent vaccines. Studies have shown that PRN is a critical component of pertussis vaccines[2]. The interaction between the mammalian cells and PRN can be specifically inhibited by an Arg-Gly-Asp (RGD)-containing synthetic peptide that is homologous with a region found between positions 939 and 1014 in the PRN gene sequence[9]. FHA is the major adhesin of B pertussis for bronchial epithelial cells[10]. Monoclonal antibodies of amino acid residues following the RGD peptide were shown to inhibit binding of B pertussis to ciliated rabbit cells in the same fashion as antibodies elicited by whole FHA[11]. In this study, mutated PTS1 and the fragments of PRN (amino acid residues 248–364) and FHA (amino acid residues 1099–1210) were used as combined and recombinant vaccines.
DNA vaccines make use of the fact that plasmid DNA can directly transfect animal cells in vivo[12]. This discovery made it possible to induce immune responses by direct injection of DNA plasmids encoding for antigenic proteins into animal cells. This method, referred to as DNA immunization, has now been used to elicit protective antibody and cell-mediated immune responses for viral, bacterial and parasitic diseases[5].
Being an extracellular pathogen, B pertussis was thought to be a humoral pathogen only, whereas evidence suggests that cellular and humoral immunity may act in concert to eliminate B pertussis infection[13]. Thus B pertussis is now considered to be a facultative intracellular pathogen. Defence against intracellular pathogens is usually dependent on the activation of a strong cellular immune response[11]. Furthermore, cellular immunity memory lasts longer than humoral immunity, and may play a role in protective immunity after infection with B pertussis[2]. DNA vaccines are simple to make and deliver, and can elicit both humoral and cellular immunity[14]. Many studies suggest that DNA vaccines induce a broad range of protective immunities, including antibodies, CD8+CTL, CD4+Th cells that challenge the pathogens[15]. Immune responses generated by DNA vaccination are initiated by professional antigen-presenting cells (APC). APC presentation of antigens in the context of major histocompatibility complexes (MHC) class I and MHC class II molecules lead to cell-mediated and humoral immunity[16]. Commonly, interleukin (IL)-4 and IL-10 are mainly elicited by Th2 cells and are typically used as markers for humoral immunity, IFN-γ and TNF-α are mainly produced by Th1 cells and are considered cellular immune[14].
The major potential disadvantage of DNA vaccination is the low immune responses. Consequently, many methods have been attempted to enhance the responses, including incorporation into liposomes, co-administration with adjuvants such as monophosphoryl lipid A, alum or co-administration of plasmids encoding cytokines[17].
In this study, we investigated the immunogenicity of a prime-boost strategy, co-injection with murine granulocyte-macrophage-colony stimulating factor (mGM-CSF) plasmids and the boost with protein for a recombinant pertussis DNA vaccine.
Materials and methods
Animals, bacterial strains and plasmids Balb/c mice (4 weeks of age) were purchased from Shanghai Laboratory Animal Co Ltd (Shanghai, China). Bordetella pertussis CS was from the Chinese National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China); plasmid pQE31, pGEX-5X-1 were from the Department of Medical Microbiology and Parasitology, Shanghai Jiao Tong University School of Medicine (Shanghai, China). These plasmids could be used for His-tag (pQE31) and GST-tag (pGEX-5X-1) purification, respectively. Plasmid pVAX1 and pCEP4/mGM-CSF (pGM-CSF) were generous gifts from Dr David D HO from the Aaron Diamond AIDS Research Center, Rockefeller University, USA and Dr Mi-hua TAO from the Institute of Biomedical Sciences, Academia Sinica (Taipei, China), respectively. Plasmid pVAX1 contains a CMV promoter allowing high-copy number replication in Escherichia coli and high-level transient expression of the protein in most mammalian cells.
PCR-based site-directed mutagenesis of PTS1 Primers P1 and P2 (see Table 1) were designed to amplify gene PTS1, and other primers P3, P4 and P5 were designed to mutate PTS1 at amino acid 9 (aa9) and aa129 following Burnette’s[7] and Loosmore’s[8] studies. PCR were conducted with Pfu DNA polymerase (Shanghai Invitrogen, Shanghai, China), synthetic oligonucleotide primers (Shanghai Invitrogen, Shanghai, China) on GeneAmp PCR System 9600 (Perkin-Elmer, Wellesley, MA, USA). PCR products were purified by Qiaquick gel extraction kit (Qiagen, Hilden, Germany).

Full table
Construction and preparation of the plasmids Primers P6 and P7 were designed to amplify the fragment of gene PRN; primers P8 and P9 were designed to amplify the fragment of gene FHA. These 2 fragments were cloned to plasmid pGEX-5X-1, as the mutated PTS1 whole gene described above was cloned to plasmid pQE31.
Primers P1, P10, P11, P12, P13 and P14 were designed to recombine gene PTS1, PRN and FHA fragments to a recombinant fragment (PPF). First, PTS1, the PRN and FHA fragments were amplified with primers P1 and P10, P11 and P12, P13 and P14, respectively. Then 3 PCR products were purified, mixed and used as templates in the next PCR reaction which used P1 and P14 as primers. There were overlaps between P10 and P11, P12 and P13, thus a full-length recombinant DNA, PTS1–PRN–FHA (PPF) could be produced.
Purified PCR products and plasmid pQE31, pGEX-5X-1, pVAX1 were digested by restriction enzymes (Takara Bio Inc., Otsu, Shiga, Japan), ligated (T4 ligase, Takara Bio Inc., Otsu, Shiga, Japan) and sequenced with ABI DNA sequencer (Perkin-Elmer, Wellesley, MA, USA). pQE31/PTS1, pQE31/PPF, pGEX-5X-1/PRN, and pGEX-5X-1/FHA were prepared for protein expression, pVAX1/PTS1, pVAX1/PRN, pVAX1/FHA and pVAX1/PPF were prepared for DNA injection with Qiagen Plasmid Mega Kit (Qiagen, Hilden, Germany).
Expression and purification of proteins Expression of proteins PTS1 and PPF: The plasmid pQE31/PTS1 and pQE31/PPF were isolated and transformed into the lac inducible expression system of E coli M15. An overnight culture was diluted 50-fold into fresh medium at 37 oC in Luria-Bertani medium containing 100 μg ampicillin (per mL) and 30 μg kanamycin (per mL). One mmol/L Isopropyl-β-D-thiogal-actopy-ranoside (IPTG) was added when the culture reached the optical density of 0.6 to 0.8 at 600 nm. The cells were harvested and resuspended in phosphate-buffered saline (PBS) 4 h later. The cell lysate containing the proteins was analyzed in SDS-PAGE and Western blotting with monoclonal antibody to 6*His-tag[18].
Protein PRN and FHA was prepared in a similar method as PTS1 and PPF, except that the expression system was E coli BL21; the concentration of IPTG was 0.15 mmol/L, and the monoclonal antibody used in the Western blotting was GST-tag.
Purification of protein PTS1 and PPF was fulfilled according to the protocol of Qiaexpressionist (Qiagen, Hilden, Germany) and protein PRN and FHA to the protocol of Glutathione Sepharose 4B (Amersham Biosciences, Little Chalfont, UK).
Animal immunization Five groups of 10 Balb/c mice were used in this study. Plasmid pVAX1/PTS1, pVAX1/PRN and pVAX1/FHA were mixed and named pVAX1/m. These 3 fragments were constructed together to a recombinant fragment PPF and then cloned in the same plasmid pVAX1 and named pVAX1/PPF. The animal immunization procedure is shown in Table 2. The volume of each injection of 50 μg was intramuscularly administered into the thigh quadriceps muscle per mouse for each plasmid or 10 μg intraperitoneally administered per mouse for protein. On d 35, serum was collected from each mouse.

Full table
ELISA assay for antigen-specific antibody production The wells of the polystyrene microdilution plates were coated overnight with 50 µL (4 µg/mL) of PT, PRN or FHA per well, respectively. The plates were washed 3 times with PBS. PBS containing 2% bovine serum albumin was used to block nonspecific binding sites for 2 h. After the plates were washed with PBS, 50 µL of serum was added to each well for 30 min at room temperature to bind to PT, PRN and FHA. After the addition of anti-murine IgG conjugated with alkaline phosphatase (Sigma-Aldrich, St Louis, MO, USA), the plates were incubated for 1 h at room temperature and then washed. The color reaction was developed by the addition of enzyme substrate p-Nitrophenyl Phosphate (Sigma-Aldrich, USA) and was quenched with the addition of 3 mol/L NaOH. The optical density was measured at 405 nm with an ELISA reader.
ELISA assay for cytokine induction The collected sera were used to detect cytokine IL-10, IL-4, IFN-γ and TNF-α with homologous ELISA kits (Boster, Wuhan, China). Serum 100 µL was added to each well and the plates were incubated for 90 min (for IL-10, IL-4 and TNF-α) or 120 min (for IFN-γ) at 37 oC. The serum was discarded from each well; 100 µL biotin-antibody reagent was added to each well, then the plates were incubated for 60 min at 37 oC and washed 3 times with PBS. With 100 µL avidin-biotin-complex reagent added to each well, the plates were incubated for another 30 min at 37 oC and washed 5 times with PBS. The plates were incubated for 20 min and protected from light after TMB reagent was added to each well. The reaction was quenched with the addition of 100 µL TMB stop reagent to each well. The optical density was measured at 450 nm with an ELISA reader.
Splenocyte-proliferation assay Lymphocytes were prepared from mouse spleens. The isolated cell suspensions were resuspended to a concentration of 2×104 cells/mL. A 100 mL aliquot containing 2×103 cells was added immediately to each well of a 96-well flat bottom microtitre plate in triplicate. rIL-2 (R&D, Minneapolis, MN, USA) and homologous proteins were added to the wells to the final concentration of 4 ng/mL (rIL-2) and 10 μg/mL (homologous proteins). Another triple well was added (rIL-2) as the control. After a period of 72 h incubation at 37 oC in 5% CO2, splenocyte-proliferation was assayed with a MTT kit (Beyotime, Haimen, China). The ratio (test/control) of the optical density at 570 nm was described as the splenocyte-proliferation value.
Statistical analysis Statistical analysis was performed using Student-Newman-Keuls test. Values were compared between different immunization groups. P values <0.05 or 0.01 were considered statistically significant.
Results
Clone, expression and purification of plasmids and antigens The gene PTS1 was mutated by a series of PCR reactions and cloned to plasmid pQE31. Then the recombinant gene PPF was constructed by PCR and cloned to plasmid pQE31; protein PTS1 and PPF were expressed and purified. Similarly, the other gene fragments, PRN and FHA were cloned to pGEX-5X-1 and the proteins were produced (Figures 1A, C, D). Proteins PTS1, PRN and FHA were purified for coating the ELISA plates used for measuring the antibody production and MTT assay, whereas the fusion protein PPF was purified for the boosts during the animal experiment.
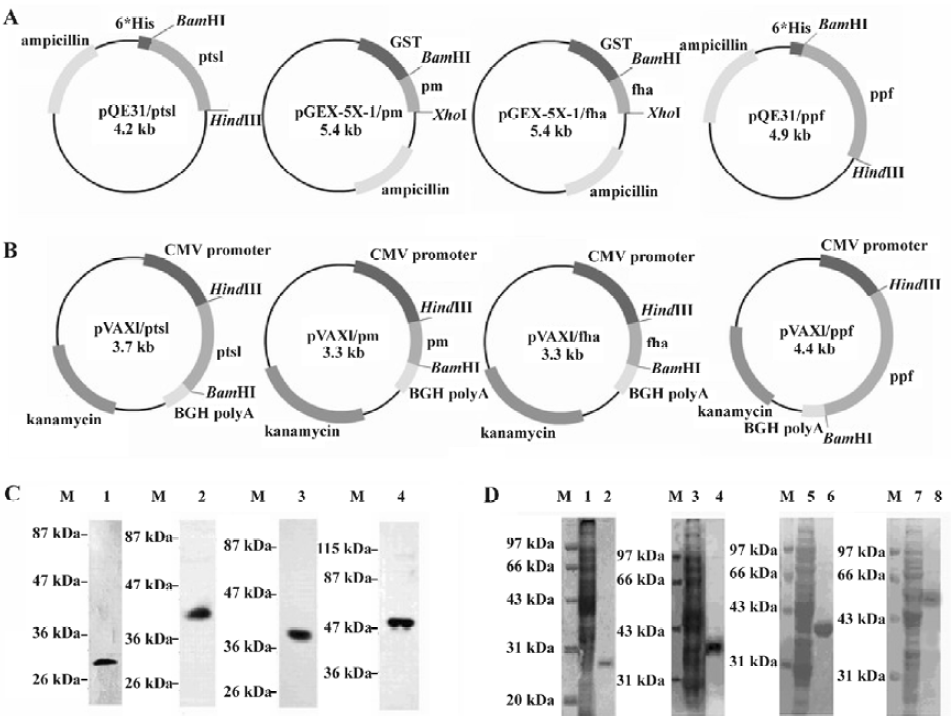
The plasmids we constructed and used in the animal immunizations are shown in Figure 1B. pVAX1/PTS1, pVAX1/PRN, pVAX1/FHA and pVAX1/PPF were digested with restriction enzyme HindIII and BamHI.
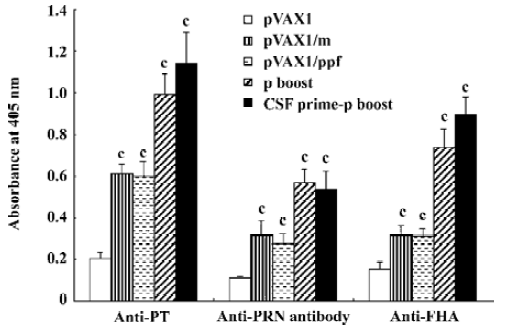
ELISA assay for antigen-specific antibody production ELISA assays were used to display antigen-specific antibody production (Figure 2). The results are similar in the 3 ELISA assays. Group pVAX1/m and group pVAX1/PPF showed more specific antibodies than control group pVAX1. The group protein boost and group CSF prime-protein boost showed a significant increase in antibodies compared to group pVAX1/m and group pVAX1/PPF. Especially in anti-PTS1 and anti-FHA, group CSF prime-protein boost showed a higher absorbance than group protein boost. These results indicate that mice boosted with protein elicited an increased humoral immune response than those boosted with DNA plasmids.
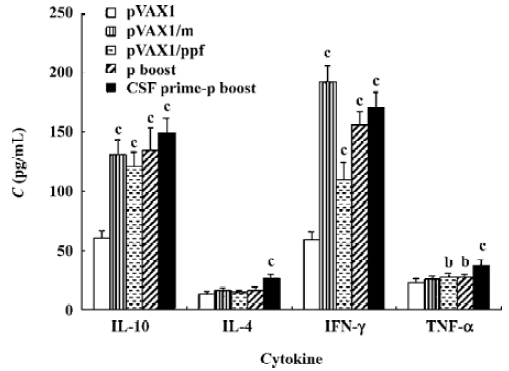
ELISA assay for cytokine induction To reveal cellular and humoral response in the mice, ELISA assays were used to describe cytokines production (Figure 3). IL-10 and IL-4 were used to indicate the level of humoral immune response, IFN-γ and TNF-α were used to reveal the level of cellular immune response. For IL-10 and IFN-γ, all the vaccine groups showed significant increases compared to the control group pVAX1. For IL-4 and TNF-α, only group CSF prime-protein boost showed more cytokine production than group pVAX1. These cytokine results show that better humoral and cellular immune responses were elicited when the mice immunized with DNA and cytokine plasmid pGM-CSF boosted with the corresponding proteins.
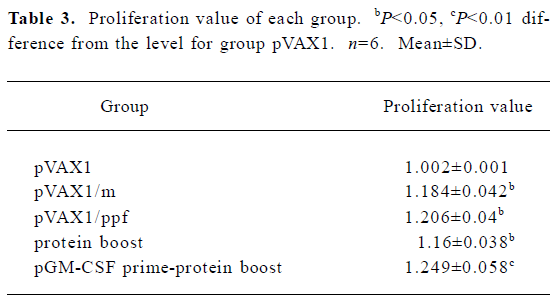
Splenocyte-proliferation assay MTT test was used to detect the splenocyte-proliferation reaction. The proliferation ratio (test/control) analyzed at 570 nm with ELISA reader are shown in Table 3. All the test groups, except the CSF prime-protein boost group, showed significant differences compared to the control group pVAX1 (P<0.05). The CSF prime-protein boost group showed a more significant difference compared to the pVAX1 group (P<0.01). There was no difference between any 2 test groups.
Full table
Discussion
B pertussis is a pathogen for humans only. Immunization is the most effective method for the prevention and control of this disease and has been used successfully for decades[19]. That primary vaccination of infants against pertussis does not always afford long-term protection suggests a need for a better immune strategy to maintain adequate levels of specific immunity to B pertussis[20].
In the present study, the mutated PTS1 gene and the 2 gene fragments described earlier were cloned into plasmid pQE31, pGEX-5X-1 for protein expression and pVAX1 for DNA immunization. The 3 plasmids (pVAX1/PTS1, pVAX1/PRN and pVAX1/FHA) were mixed as a mixed vaccine. These 3 genes were also constructed to a recombinant fragment PPF, and then constructed to another DNA plasmid pVAX1/PPF.
There are several benefits of multiple antigens being encoded as DNA vaccines. For example, different components may have different functions. Another advantage of using multiple genes is the ability to target more than one stage of the life cycle of a pathogen[14]. For pertussis, the first stage of the cycle is the bacterium entering the host via the airways, adhering to ciliated epithelial cells in the trachea and nasopharynx with FHA and PRN actions. In the next stage, PT and other toxins are secreted by the micro-organism damaging the epithelial lining and resulted in the loss of ciliated cells which induce symptoms[11]. In the work described here, either the mixed plasmids or the recombinant one can elicit specific antibodies: anti-PTS1, anti-PRN and anti-FHA. These antigen-specific antibodies may play a role in protection against B pertussis in these different stages.
In the cytokine assays, all of the pertussis DNA recombinant plasmids elicited IL-10, IFN-γ and specific antibodies. The assay showed that these plasmids can elicit both humoral immunity and cellular immunity. The relatively poor efficacy of DNA vaccines in immune responses, especially in large animals, has limited their practical use. Here, we provide a strategy to improve the immune response induced by DNA vaccines by supplementing it with the GM-CSF expression vector and boosting it with protein antigens.
It is well known that parenterally-delivered DNA vaccines do not efficiently activate specific secreted antibody responses at mucosal sites, further reducing the applicability of effective vaccines against mucosal bacterial pathogens[21]. However, with a boost, the systemic immune response can be largely improved[22]. In this study, a recombinant protein in adjuvant was used as a boosting agent and significantly enhanced immune response. All the antibodies elicited in the group protein boosted showed significant increases than those in group non protein boosted.
Another solution to improve the immune response of DNA vaccines is the co-injection of cytokine plasmids. Cytokines have been used as genetic adjuvants to enhance the efficacy of DNA immunization. The co-injection of cytokine genes for DNA vaccines can modulate antigen-specific immune responses and augment immune response[23]. GM-CSF is produced by T and B cells, endothelial cells, fibroblasts and macrophages. Although the mechanism through which GM-CSF enhances immunity is not completely understood, evidence suggests that GM-CSF may work on APC[24]. GM-CSF increases expression of major histocompatibility complex class II molecules, enhances antigen presentation, and augments antigen-specific T-cell proliferative response[25]. Thus GM-CSF expression plasmid plays an important role in the amplification of humoral and cell-mediated immune responses[26]. In this study, GM-CSF plasmid was co-injected with recombinant fragment plasmids to elicit a better immune response.
Specific antibodies and IL-4 and IL-10 were detected to investigate humoral immune responses in this study. For specific antibodies, the specific antibodies in group protein boost and group CSF prime-protein boost were significantly increased compared to the other groups. For IL-10, all 4 test groups showed significant increases compared to the control group pVAX1, and there was no significant difference between any 2 test groups. For IL-4, only the CSF prime-protein boost group showed significant increase compared to the control group. Hence, only in the CSF prime-protein boost group were all the antibodies and cytokines significantly different compared to the pVAX1 group, which indicates that when primed with pGM-CSF and boosted with homologous proteins, a much more enhanced humoral immune response was produced.
In the present study, splenocyte-proliferation assay along with IFN-γ and TNF-α detection was used to describe cellular immune responses. The proliferation ratio from the splenocyte-proliferation assay was used to analyze the specific spleen T cell proliferation. All the test groups showed increases compared to the control. Only the CSF prime-protein boost group elicited a significant increase with similar conditions in the TNF-α and IL-4 assay. However, for IFN-γ, the CSF prime-protein boost group showed a significant increase compared to the control groups and group pVAX1/PPF, but IFN-γ produced in group CSF prime-protein boost was less than those in the mixed DNA plasmids group pVAX1/m. Indeed, group CSF prime-protein boost is the only group where all parameters showed a significant increase compared to the pVAX1 group, indicating that an enhanced cellular immune response was elicited with the GM-CSF plasmid prime-protein boost.
Usually DNA vaccination is particularly effective in priming an immune response, but the low amount of protein antigens synthesized in the host limited this type of immunization[27]. Otherwise, DNA prime-protein boost immunization can enhance both antigen-specific antibody and Th1-type cellular immune responses in other studies[28]. Many cytokines, such as IL-2[15,29,30], IFN-γ[17], GM-CSF[29], IL-12[15,31] have been co-injected to provoke better immune and protection responses. In our experiment, we combined these 2 strategies and demonstrated the increased immunogenicity of a recombinant pertussis DNA vaccine.
In conclusion, a multiple antigen-recombinant pertussis DNA vaccine can elicit the homologous specific protection antibodies and cellular immune response. When the recombinant DNA plasmid is co-injected with another plasmid pGM-CSF as the prime, and boosted with homologous purified protein, it showed significant increased specific humoral and cellular immune responses. The results reported here provide a useful strategy of DNA immunization and may be applied in the future.
Acknowledgements
We are extremely grateful to Dr David D HO (Rockefeller University, USA) and Dr Mi-Hua TAO (Institute of Biomedical Sciences, Academia Sinica, China) for kindly providing the plasmids. We also thank Dr Jia-yan CHEN (University of Southern California, USA) for the critical reading of the manuscript.
References
- Franco E, Giambi C, Ialacci R, Maurici M. Pertussis vaccination for adolescents and adults. Expert Opin Biol Ther 2004;4:1669-76.
- Pichichero ME, Casey JR. Acellular pertussis vaccines for adolescents. Pediatr Infect Dis J 2005;24:S117-26.
- Kamachi K, Arakawa Y. Expression of a C terminally truncated form of pertussis toxin S1 subunit effectively induces protection against pertussis toxin following DNA-based immunization. Infect Immun 2004;72:4293-6.
- Halperin SA. Pertussis–a disease and vaccine for all ages. N Engl J Med 2005;353:1615-7.
- Kamachi K, Konda T, Arakawa Y. DNA vaccine encoding pertussis toxin S1 subunit induces protection against Bordetella pertussis in mice. Vaccine 2003;21:4609-15.
- Bassinet L, Fitting C, Housset B, Cavaillon JM, Guiso N. Bordetella pertussis adenylate cyclase-hemolysin induces interleukin-6 secretion by human tracheal epithelial cells. Infect Immun 2004;72:5530-3.
- Burnette WN, Cieplak W, Mar VL, Kaljot KT, Sato H, Keith JM. Pertussis toxin S1 mutant with reduced enzyme activity and a conserved protective epitope. Science 1988;242:72-4.
- Loosmore SM, Zealey GR, Boux HA, Cockle SA, Radika K, Fahim RE, et al. Engineering of genetically detoxified pertussis toxin analogs for development of a recombinant whooping cough vaccine. Infect Immun 1990;58:3653-62.
- Charles IG, Dougan G, Pickard D, Chatfield S, Smith M, Novotny P, et al. Molecular cloning and characterization of protective outer membrane protein P.69 from Bordetella pertussis. Proc Natl Acad Sci USA 1989;86:3554-8.
- Belcher CE, Drenkow J, Kehoe B, Gingeras TR, McNamara N, Lemjabbar H, . The transcriptional responses of respiratory epithelial cells to reveal host defensive and pathogen counter-defensive strategies. Proc Natl Acad Sci USA 2000; 97: 13 847–52.
- Smith AM, Guzman CA, Walker MJ. The virulence factors of Bordetella pertussis: a matter of control. FEMS Microbiol Rev 2001;25:309-33.
- Wolff JA, Malone RW, Williams P, Chong WG, Acsadi A, Jani A, et al. Direct gene transfer into mouse muscle in vivo. Science 1990;247:1465-8.
- Heininger U. Recent progress in clinical and basic pertussis research. Eur J Pediatr 2001;160:203-13.
- Doria-Rose NA. Haigwood NL. DNA vaccine strategies: candidates for immune modulation and immunization regimens. Methods 2003;31:207-16.
- Du DW, Jia ZS, Li GY, Zhou YY. HBV DNA vaccine with adjuvant cytokines induced specific immune responses against HBV infection. World J Gastroenterol 2003;9:108-11.
- Leachman SA, Tigelaar RE, Shlyankevich M, Slade MD, Irwin M, Chang E, et al. Granulocyte-macrophage colony-stimulating factor priming plus papillomavirus E6 DNA vaccination: effects on papilloma formation and regression in the cottontail rabbit papillomavirus–rabbit model. J Virol 2000;74:8700-8.
- Nimal S, McCormick AL, Thomas MS, Heath AW. An interferon gamma-gp120 fusion delivered as a DNA vaccine induces enhanced priming. Vaccine 2005;23:3984-90.
- Liu DF, Phillips E, Wizemann TM, Siegel MM, Tabei K, Cowell JL, et al. Characterization of a recombinant fragment that contains a carbohydrate recognition domain of the filamentous hemagglutinin. Infect Immun 1997;65:3465-8.
- He Q, Makinen J, Berbers G, Mooi FR, Viljanen MK, Arvilommi H, et al. Bordetella pertussis protein pertactin induces type-specific antibodies: one possible explanation for the emergence of antigenic variants? J Infect Dis 2003;187:1200-5.
- Carter CR, Dagg BM, Whitmore KM, Keeble JR, Asokanathan C., Xing D, et al. The effect of pertussis whole cell and acellular vaccines on pulmonary immunology in an aerosol challenge model. Cell Immunol 2004;227:51-8.
- Lasaro MO, Luiz WB, Sbrogio-Almeida ME, Ferreira LC. Prime-boost vaccine regimen confers protective immunity to human-derived enterotoxigenic Escherichia coli. Vaccine 2005;23:2430-8.
- Mielcarek N, Cornette J, Schacht AM, Pierce RJ, Locht C, Capron A, et al. Intranasal priming with recombinant Bordetella pertussis for the induction of a systemic immune response against a heterologous antigen. Infect Immun 1997;65:544-50.
- Kim JJ, Yang JS, Dentchev T, Dang K, Weiner DB. Chemokine gene adjuvants can modulate immune responses induced by DNA vaccines. J Interferon Cytokine Res 2000;20:487-98.
- Sun X, Hodge LM, Jones HP, Tabor L, Simecka JW. Co-expression of granulocyte-macrophage colony-stimulating factor with antigen enhances humoral and tumor immunity after DNA vaccination. Vaccine 2002;20:1466-74.
- Kamath AT, Hanke T, Briscoe H, Britton WJ. Co-immunization with DNA vaccines expressing granulocyte-macrophage colony-stimulating factor and mycobacterial secreted proteins enhances T-cell immunity, but not protective efficacy against Mycobacterium tuberculosis. Immunology 1999;96:511-6.
- Sin JI., Sung JH, Suh YS, Lee AH, Chung JH, Sung YC. Protective immunity against heterologous challenge with encephalo-myocarditis virus by VP1 DNA vaccination: effect of coinjection with a granulocyte-macrophage colony stimulating factor gene. Vaccine 1997;15:1827-33.
- Tanghe A, D’Souza S, Rosseels V, Denis O, Ottenhoff TH, Dalemans W, et al. Improved immunogenicity and protective efficacy of a tuberculosis DNA vaccine encoding Ag85 by protein boosting. Infect Immun 2001;69:3041-7.
- Wang QM, Sun SH, Hu ZL, Yin M, Xiao CJ, Zhang JC. Improved immunogenicity of a tuberculosis DNA vaccine encoding ESAT6 by DNA priming and protein boosting. Vaccine 2004;22:3622-7.
- Bharati K, Appaiahgari MB, Vrati S. Effect of cytokine-encoding plasmid delivery on immune response to Japanese encephalitis virus DNA vaccine in mice. Microbiol Immunol 2005;49:349-53.
- He X, Tsang TC, Zhang T, Luo P, Harris DT. Antigen epitope-expressing cytokines for DNA immunization. Vaccine 2005;23:1966-72.
- Chattergoon MA, Saulino V, Shames JP, Stein J, Montaner LJ, Weiner DB. Co-immunization with plasmid IL-12 generates a strong T-cell memory response in mice. Vaccine 2004;22:1744-50.