
Anti-inflammatory effects of a novel peptide designed to bind with NF-κB p50 subunit1
Introduction
As a ubiquitous transcription factor, NF-κB is one of the cross-talk points of multiple signal transduction pathways, which plays a key role in the regulation of transcription and the expression of many genes such as cytokines, adhesion molecules, chemotaxins, acute phase response proteins and enzymes; it also participates in basic processes such as cell proliferation, virus replication, inflammatory responses, apoptosis and immune responses[1,2]. Excess activation of NF-κB is associated with processes of various inflammatory diseases such as allergy, asthma, autoimmune diseases, atherosclerosis, graft rejection, enteronitis, cachexia, rheumatoid arthritis, etc[3–9]. These days, many scientists suggest that NF-κB may become a potential target for the therapy of inflammatory diseases[10–14]. Therefore, we designed short peptides against the NF-κB p50 subunit with bioinformatics technique to antagonize NF-κB activity.
The purpose of the present study was to identify the anti-inflammatory functions of 1 peptide designed to bind with the NF-κB p50 subunit with in vitro and in vivo models.
Materials and methods
Animals Inbred BALB/c mice (20–25 g) were purchased from Chongqing Medical University (Chongqing, China) and maintained on a standard chow pellet diet with pure water ad libitum and a 12:00 h light/dark cycle.
Materials The NF-κB p50 subunit was purchased from the Active Motif Company (Carlsbad, CA, USA). Chemicals such as phorbol myristate acetate, zymosan A, lipopolysac-charide, etc, were from Sigma-Aldrich Chemical (St Louis, MO, USA). Fetal calf serum and Roswell Park Memorial Institute (RPMI)-1640 medium were purchased from Hyclone (Logan, UT, USA). The THP-1 cell line was from ATCC (Manassas, VA, USA). The ELISA kits for tumor necrosis factor-α (TNF-α) and interleukin 6 (IL-6) were purchased from Jingmei Biotech (Shenzhen, China). The anti-inflammatory peptide (AIP) is a fusion peptide with the sequence GRKKRRQRRRPPQ (cell-penetrating peptide)-CPVIRH (p50-binding peptide). The AIP and the negative control peptide (NCP; sequence: GRKKRRQRRRPPQ–EGTRKN) were designed by Dr Xiang XU (State Key Laboratory of Trauma, Burns and Combined Injury, Department 1, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China) using Insight II soft package (Accelrys, San Diego, CA, USA) with the multiple copy simultaneous search (MCSS) program and synthesized by Sangon Company (Shanghai, China).
Affinity between the AIP and the NF-κB p50 subunit A IAsys plus biosensor from Thermo Labsystems (Helsinki, Finland) was used to measure the affinity of AIP binding with p50 by means of kinetic analysis according to manufac-turer’s instructions. The value of the KD (dissociation equilibrium constant) of AIP binding with p50 was calculated with FASTfit software (Thermo Labsystems, Helsinki, Finland).
Phorbol myristate acetate (PMA)-induced ear edema PMA and dexamethasone (Dex) were dissolved in ethanol and 0.9% NaCl at concentrations of 250 µg/mL and 2 mg/mL, respectively. The AIP was dissolved in 0.9% NaCl at concentrations of 0.01 mg/mL, 0.1 mg/mL, 1 mg/mL, 2 mg/mL and 4 mg/mL. Each mouse received 5 µg/ear of PMA on the left ear and 20 µL ethanol on the right ear[15]. Dex, 0.9% NaCl, or different doses of the AIP were given on the left ear (20 µL/ear, respectively) 15 min after treatment with PMA; the right ear received 0.9% NaCl at the same time. This practice was applied to both the inner and outer surfaces of the ear. According to the results of Carlson et al[16], we measured the thickness of the ears with a microgauge 6 h after PMA application, and the increase in the thickness of mice ears-thickness of left ear-thickness of right ear.
Histological examination The ears of the mice were collected and fixed in 10% neutral formalin and stored at 4 oC for later processing. After being washed, dehydrated and embedded in paraffin, the ear sections (6-µm thickness) were stained with Mayer’s hematoxylin and eosin (H&E) for assessment of tissue damage and inflammatory cell infiltration. H&E stained sections were examined by a light microscope and digital camera system (Olympus Optical, Tokyo, Japan).
Zymosan A-induced peritonitis Measurements of the number of inflammatory cells was performed by the methods described by Ajuebor et al[17] and Getting et al[18]. Briefly, the mice were injected intraperitoneally concomitantly with zymosan A (0.4 mg/mL) and either Dex (100 µg/mL) or the designated doses of the AIP in 1 mL of saline. The animals were sacrificed by inhalation of CO2 at the 4 h time-point for counting peritoneal polymorphonuclear granulocytes (PMN) and monocytes plus macrophages (mono-Mø), respectively. The peritoneal cells were collected by lavage with 3 mL of 0.1 mol/L phosphate buffered solution containing 3 mmol/L ethylenediaminetetraacetic acid. Aliquots of the lavage fluid were stained with Turk’s solution (0.01% crystal violet in 3% acetic acid) and differential cell counts were performed using a disposable hematocytometer and a light microscope (Olympus Optical, Tokyo, Japan). The lavage fluids were centrifuged at 400×g for 10 min and supernatants stored at -20 °C before evaluating TNF-α and IL-6 levels by ELISA.
Cytokines THP-1 cells (2×105/mL) were incubated in 24-well plates (0.5 mL/well) for 24 h (37 °C, 5% CO2) with 20 ng/mL PMA; the supernatants were then rejected. The cells were washed twice in 10% fetal calf serum-RPMI 1640, and cocultured with different doses of the AIP in the presence or absence of 100 ng/mL lipopolysaccharide (LPS) for 6 h (37 °C, 5% CO2). After incubation, the concentrations of TNF-α and IL-6 in the supernatant were determined by ELISA kits.
Statistical analysis Data were presented as mean±SD for each group. Student’s t-test was used to determine significant differences. The critical level for significance was set at P<0.05.
Results
Affinity between the AIP and the NF-κB p50 subunit Kinetic analysis with biosensor technology demonstrated that the AIP, not the NCP, can really interact with p50 (Figure 1), and the Kd of AIP binding with p50 was 2.77×10-6 mol/L (Table 1).
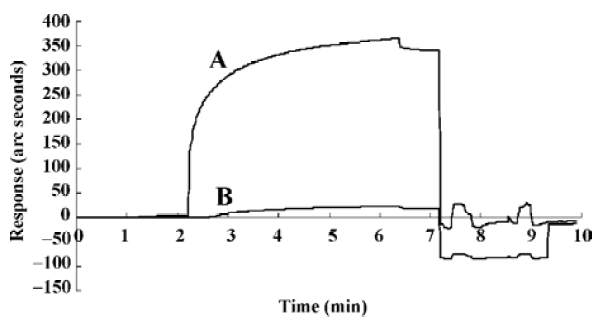
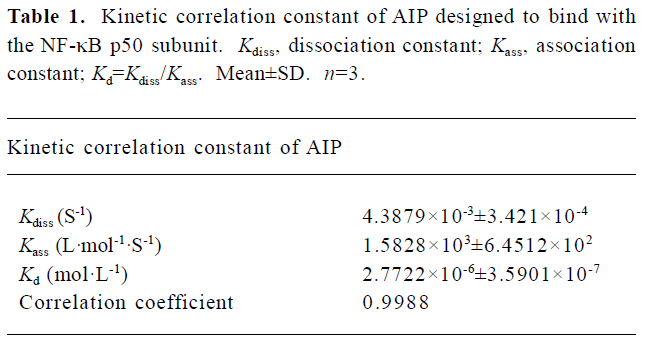
Full table
Effects of the AIP on PMA-induced ear edema in mice The inhibitory effect of the AIP on ear edema was dose-dependent from 0.2 µg/ear to 2 µg/ear (Table 2). We observed that the degree of ear edema in the group of 2 µg/ear decreased significantly compared with that of the vehicle group (0.9% NaCl; P<0.01), and the mean inhibition of this group reached 50%. However, the inhibitory effect was reduced when the dose of the AIP was higher than 2 µg/ear. Pathological sections demonstrated that the AIP could remarkably decrease inflammatory cell infiltration of PMA-induced ear edema tissue (Figure 2). The NCP had no significantly inhibitory effects on ear edema compared with the vehicle group.
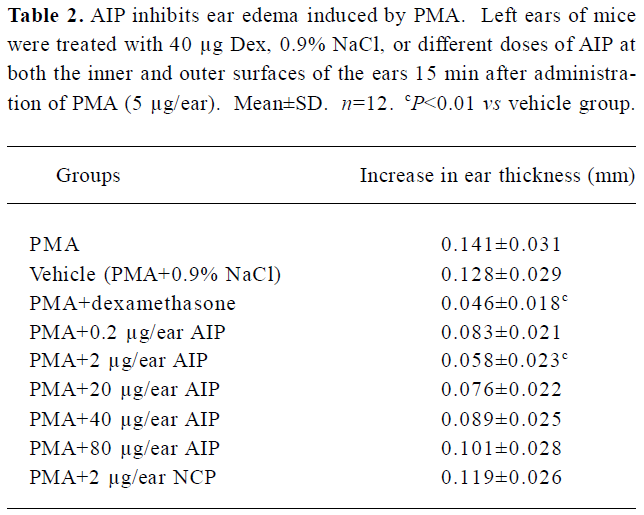
Full table
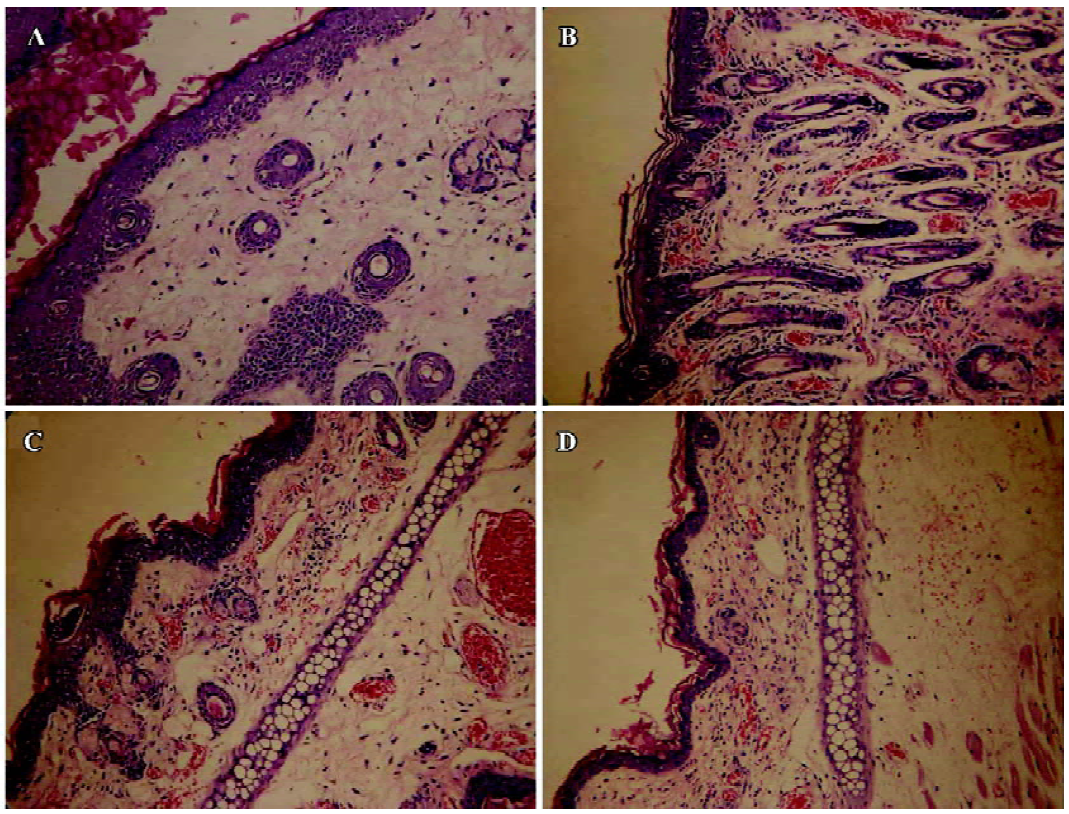
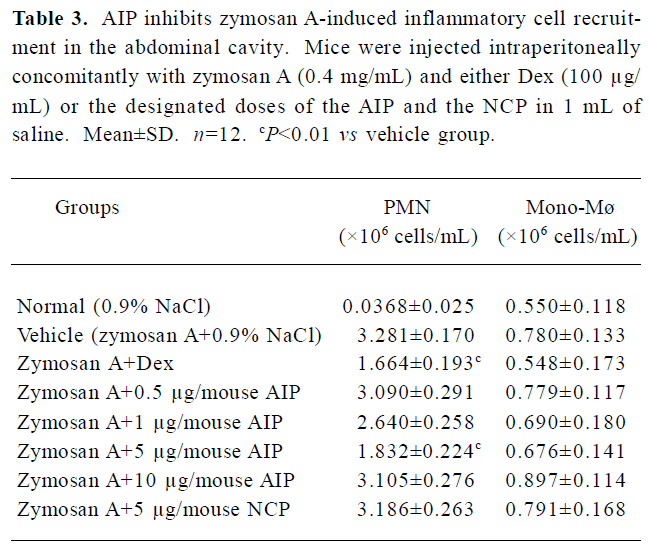
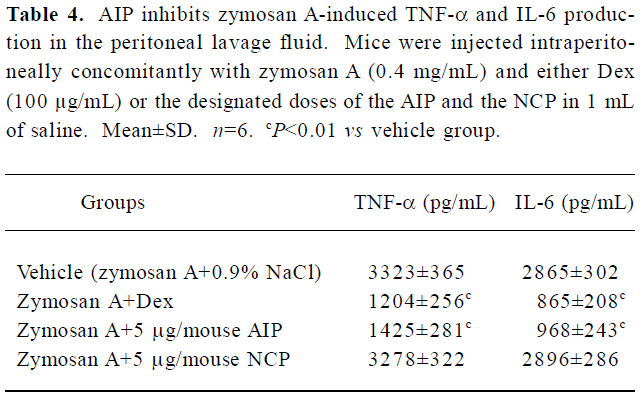
Effects of the AIP on zymosan A-induced peritonitis in mice The AIP can inhibit zymosan A-induced inflammatory cell recruitment dose-dependently from 0.5 µg/mouse to 5 µg/mouse (Table 3). The number of PMN in the group of 5 µg/mouse declined significantly compared with that of the vehicle group (0.9% NaCl; P<0.01), but the number of PMN increased when the dose of the AIP was higher than 5 µg/mouse. There was no remarkable difference in the number of mono-Mø in all the groups. In addition, the AIP can significantly inhibit zymosan A-induced TNF-α and IL-6 production in the peritoneal lavage fluid compared with the vehicle group (P<0.01, Table 4). The NCP had no obvious inhibitory effects on the number of inflammatory cells and levels of cytokines compared with the vehicle group.
Full table
Full table
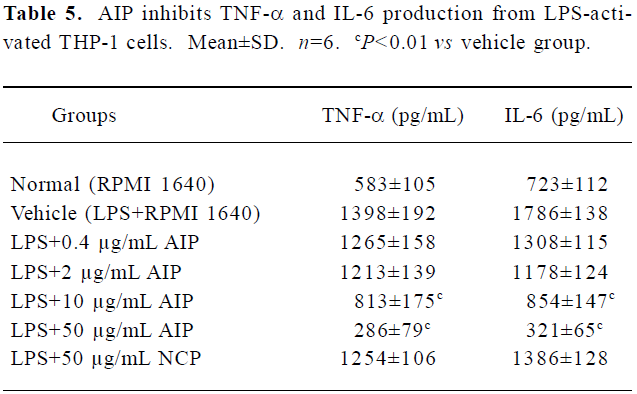
Effects of the AIP on TNF-α and IL-6 levels from LPS-treated THP-1 cells The AIP can dose-dependently inhibit pro-inflammatory cytokine TNF-α and IL-6 production from LPS-activated THP-1 cells (Table 5). TNF-α and IL-6 levels in the group of 10 and 50 μg/mL decreased significantly compared with that of vehicle group (RPMI 1640; P<0.01); the NCP has no remarkably inhibitory effect on these cytokines levels compared with the vehicle group.
Full table
Discussion
Inflammation results from the activation of many cytokines, receptors and signal pathways which form a complex network and waterfall effects, thus, it is scarcely effective to develop antagonists against single mediators of inflammation such as IL-1, IL-6, IL-8, TNF-α and so on[10]. NF-κB is a cross-talk point of these multiple signal transduction pathways, and its activation by pro-inflammatory stimuli leads to increased expression of many genes involved in inflammation. Therefore, it is crucial for the treatment of inflammation to selectively inhibit NF-κB activation induced by pro-inflammatory factors[15]. In this study, we provide evidence for anti-inflammatory effects of a novel peptide designed to bind with the NF-κB p50 subunit. The present study reveals that the AIP possessed the ability to relieve local acute inflammation, and the mechanisms by which the AIP inhibits inflammation may be the inhibition of NF-κB activation by binding the NF-κB p50 subunit with the AIP. Certainly, it is not enough to identify its molecular mechanism of anti-inflammatory action by biosensor technology and cytokine detection in vitro. In order to confirm its mechanisms, further studies need to be done, such as NF-κB responsive luciferase reporter assay, quantitative PCR methods, electrophoretic mobility shift assay, etc.
The effects of the AIP in vivo were tested in 2 distinct experimental mouse models of acute inflammation. The results suggest that the AIP is an effective inhibitor of inflammation in these experimental models. However, when the dose of the AIP was higher than the optimal dose, there were toxic side effects instead of anti-inflammatory effects. We observed that there were many dead cells in the peritoneal lavage fluid when the dose of the AIP was 10 μg/mouse. According to the fact that NF-κB regulates the expression of genes not only for immune and inflammatory responses, but also for cell proliferation and apoptosis[8,9], NF-κB plays important roles in sustaining defense function and life of cells, and excessive inhibition of NF-κB results in apoptosis of liver cells, decreased immune function and increased sensitivity to infection[19]. Thus, high doses of the AIP leads to excessive inhibition of NF-κB which causes massive death of inflammatory cells and normal cells, and these dead cells can become new sources to induce additional inflammation. Therefore, high doses of the AIP has no anti-inflammatory effects and will cause toxic side effects.
In conclusion, we have identified the anti-inflammatory effects of AIP with some in vitro and in vivo experiments. It may have therapeutic potential for the treatment of local acute inflammation and its anti-inflammatory action will be explored on some other inflammatory models with different species.
References
- Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649-83.
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol 1994;10:405-55.
- Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell 1996;87:13-20.
- Ganchi PA, Sun SC, Greene WC, Ballard DW. I kappa B/MAD-3 masks the nuclear localization signal of NF-kappa B p65 and requires the transactivation domain to inhibit NF-kappa B p65 DNA binding. Mol Biol Cell 1992;3:1339-52.
- Parry GC, Mackman N. A set of inducible genes expressed by activated human monocytic and endothelial cells contain kappa B-like sites that specifically bind c-Rel-p65 heterodimers. J Biol Chem 1994; 269: 20 823–5.
- Ernst MK, Dunn LL, Rice NR. The PEST-like sequence of I kappa B alpha is responsible for inhibition of DNA binding but not for cytoplasmic retention of c-Rel or RelA homodimers. Mol Cell Biol 1995;15:872-82.
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 2000;18:621-63.
- Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell 1995;80:529-32.
- Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994;78:773-85.
- Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol 2005;5:435-45.
- Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell 2002;9:625-36.
- Latimer M, Ernst MK, Dunn LL, Drutskaya M, Rice NR. The N-terminal domain of IkappaB alpha masks the nuclear localization signal(s) of p50 and c-Rel homodimers. Mol Cell Biol 1998;18:2640-9.
- Johnson C, van Antwerp D, Hope TJ. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J 1999;18:6682-93.
- Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell 1998;95:759-70.
- May MJ, D’Acquisto F, Madge LA, Glöckner J, Pober JS, Ghosh S. Selective Inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science 2000;289:1550-4.
- Carlson RP, O’Neill-Davis L, Chang J, Lewis AJ. Modulation of mouse ear edema by cyclooxygenase and lipoxygenase inhibitors and other pharmacologic agents. Agents Actions 1985;17:197-204.
- Ajuebor MN, Virag L, Flower RJ, Perretti M, Szabo C. Role of inducible nitric oxide synthase in the regulation of neutrophil migration in zymosan-induced inflammation. Immunology 1998;95:625-30.
- Getting SJ, Flower RJ, Perretti M. Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br J Pharmacol 1997;120:1075-82.
- Wang CY, Guttridge DC, Mayo MW, Baldwin AS Jr. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol 1999;19:5923-9.