
Altered pharmacokinetics of zalcitabine by concurrent use of NSAIDs in rats1
Introduction
Zalcitabine (2',3'-dideoxycytidine; ddC), a pyrimidine nucleoside, is highly active against human immunodeficiency virus (HIV) and also hepatitis B virus (HBV)[1–3]. It has been used for the treatment of HIV infection and related diseases in combination with other antiretroviral agents[4,5]. Zalcitabine is rapidly and extensively absorbed after oral administration, and the primary route of elimination is renal excretion of unchanged drug, with 60% –70% of an oral dose recovered in urine within 24 h[5,6]. Renal clearance of zalcitabine exceeds the glomerular filtration rate, implying that the drug undergoes active tubular secretion in the kidney[6,7].
Zalcitabine has been reported to interact with organic anion transporters[8,9]. Among the isoforms of organic anion transporters expressed in several organs and tissues, including kidney, liver and brain, OAT1 and OAT3 play a pivotal role in the renal excretion of a wide variety of important therapeutics, including β-lactam antibiotics, diuretics, hippurates and nucleoside antiviral drugs[8–13]. Given that a considerable number of drugs and toxins can interact with organic anion transporters, potential drug interactions via OAT-mediated renal excretion may require close monitoring during combination therapies involving zalcitabine. Particular attention should be paid to interactions with commonly prescribed or over-the-counter drugs that could affect the efficacy and toxicity of zalcitabine. Furthermore, considering the short plasma half-life of zalcitabine (1–3 h), the inhibition of renal excretion of zalcitabine by the concomitant use of OAT inhibitors may prolong systemic exposure to zalcitabine, resulting in less frequent dosing. Therefore, in the present study we aimed to investigate the pharmacokinetic interactions between zalcitabine and NSAIDs (OAT inhibitors) in rats. Because rats and humans share high sequence homology for OAT1 (9%) and OAT3 (79%)[10], the rat was selected as an animal model for our pharmacokinetic studies of zalcitabine.
Materials and methods
Materials Zalcitabine, naproxen, ketoprofen, and 5-bromo-2' -deoxyuridine (BDU) were purchased from Sigma (St Louis, MO, USA). All other chemicals were of analytical grade and all solvents were of high performance liquid chromatography (HPLC) grade.
Animal studies All animal studies were performed in accordance with the experimental protocols approved by the Animal Care Committee of Chosun University. Male Sprague-Dawley rats weighing 280–300 g were obtained from Samtako Bio Co (Osan, Korea). Rats were divided into 3 groups, comprising 5 rats each group. Groups 1–3 were given an iv injection of zalcitabine (20 mg/kg) with either (1) naproxen sodium (20 mg/kg) or (2) ketoprofen (20 mg/kg) 30 min prior to the administration of zalcitabine, or (3) no concomitant treatment (control). Blood samples were collected from the right femoral artery at 0, 0.083, 0.16, 0.33, 0.5, 1, 2, 4, 8, 12, and 24 h following the zalcitabine administration. Urine was also collected at 0, 8, 12, and 24 h post zalcitabine administration from the same group of rats. Blood samples were centrifuged at 3 000 r/min for 10 min to obtain plasma for the HPLC assay. Urine samples were centrifuged at 3 000 r/min for 10 min and then passed through a membrane filter (0.45 µm). All samples were stored at -70 °C until analysis.
HPLC assay The plasma and urine concentrations of zalcitabine were determined by an HPLC assay modified from the method of Ibrahim and Boudinot[14]. Briefly, the internal standard (20 µg/mL of BDU) was added to plasma samples and then samples were deproteinized by adding acetonitrile. After centrifugation of the samples at 3 000 r/min for 10 min, the supernatant was completely evaporated with nitrogen stream. The residue was reconstituted with 100 µL of the mobile phase, and then 50 µL aliquots were injected directly into the HPLC system. The filtered urine samples (50 µL) were injected into the HPLC system after appropriate dilution. The chromatographic system consisted of a pump (LC-10AD), an automatic injector (SIL-10A) and a UV detector (SPD-10A; Shimadzu Scientific Instruments, Japan) set at 254 nm. An octadecylsilane column (Gemini C18, 4.6 mm×250 mm, 5 µm; Phenomenex, Torrance, CA, USA) was eluted with a mobile phase consisting of 10% methanol in phosphate buffer (pH 6.8) at a flow rate of 1.0 mL/min. The calibration curve from the standard samples was linear over the concentration range of 0.1 µg/mL to 20 µg/mL. The intra-day (n=5) and inter-day (n=5) coefficients of variation were less than 5%. The limit of detection was 0.1 µg/mL.
Pharmacokinetic analysis Non-compartmental pharmacokinetic analysis was performed using Kinetica-4.3 (InnaPhase Corp, Philadelphia, PA, USA). The area under the plasma concentration-time curve (AUC) was calculated by using the linear trapezoidal method. The terminal elimination rate constant (λz) was estimated from the slope of the terminal phase of the log plasma concentration-time points fitted by the method of least-squares, and then the terminal elimination half-life (T1/2) was calculated as 0.693/λz. Total clearance (CL) was estimated by dividing dose by AUC and the renal clearance (CLR) was determined as CLR=Ae/AUC, where Ae (amount of unchanged drug eliminated in urine) and AUC were measured over the same time interval.
Statistical analysis Data are expressed as mean±SD, and analyzed using one-way analysis of variance (ANOVA), followed by a posteriori testing with use of the Dunnett correction. P<0.05 was considered statistically significant.
Results
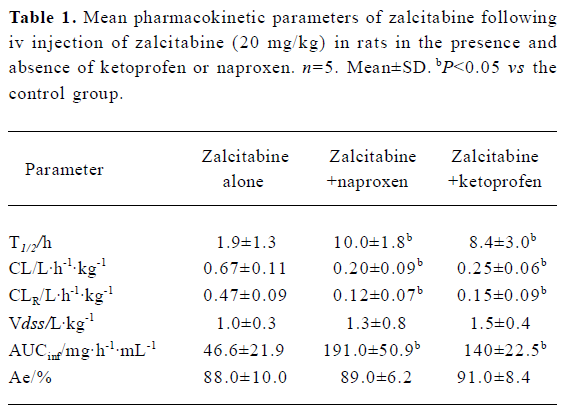
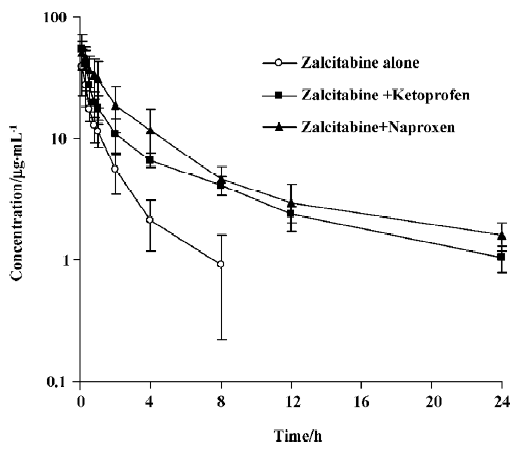
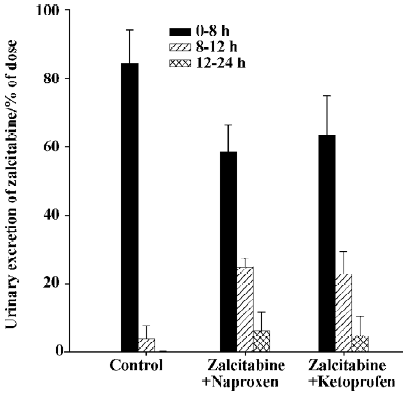
As summarized in Table 1, pretreatment with naproxen 30 min prior to zalcitabine administration significantly (P<0.05) altered the pharmacokinetics of zalcitabine in rats, compared with the controls given zalcitabine alone. Renal clearance of zalcitabine accounted for approximately 70% of the CL in all cases, which is consistent with previous reports[14]. However, renal clearance and total clearance of zalcitabine decreased by approximately 3–4-fold in the presence of naproxen or ketoprofen. Consequently, the AUC of zalcitabine was significantly (P<0.05) greater than that for the controls given zalcitabine alone (Table1, Figure 1). The terminal plasma half-life (T1/2) of zalcitabine increased by 4–5-fold in the presence of naproxen or ketoprofen. In the control group, urinary excretion of zalcitabine was rapid, and approximately 84% of the dose was excreted into urine within the first 8 h. Following the co-administration of naproxen, the urinary excretion of zalcitabine was 58%, 25%, and 6% of the administered dose in the 8 h, 12 h and 24 h urine samples, respectively (Figure 2). In the presence of ketoprofen, the urinary excretion of zalcitabine was similar to that observed with the co-administration of naproxen.
Full table
Discussion
In addition to causing changes in drug metabolism, particularly via cytochrome P450-mediated metabolism, there is increasing evidence suggesting that modulation of drug transporters can cause clinically important drug interactions. For example, the bioavailability and the intracellular concentrations of protease inhibitors can be increased in the presence of potent P-gp inhibitors[15,16]. For cationic drugs, decreases in the renal excretion of dofetilide and procainamide by the co-administration of cimetidine can be explained by the inhibition of organic cation transporter-mediated active secretion in the basolateral membranes of renal proximal tubules[17,18]. Although the majority of drug interactions have the potential to cause adverse effects, some interactions mediated by organic anion transporters have a positive impact, for example combination therapy with cidofovir and probenecid, in which the probenecid significantly reduces the nephrotoxicity of cidofovir[19]. Therefore, transport proteins can play an important role in many clinical drug interactions, either negatively or positively.
The organic anion transporter family has been implicated in the distribution of zalcitabine to the central nervous system and the proximal tubular cells in the kidney[8,9]. Recently, Khamdang et al also found that NSAIDs such as ketoprofen, indomethacin, diclofenac, naproxen, and ibuprofen inhibit organic anion uptake mediated by organic anion transporters[12]. In particular, ketoprofen and naproxen appear to be potent inhibitors of OAT1 and OAT3 located in the basolateral side of the renal tubular cells. Therefore, in our study, ketoprofen and naproxen were chosen to investigate potential drug interactions with zalcitabine. Because plasma concentrations of zalcitabine decline very rapidly in rats, larger doses must be administered to adequately characterize the behavior of zalcitabine, as reported in previous pharmacokinetic studies[7,14]. Thus, in the present study, the dose of zalcitabine administered to rats was relatively large in comparison to the doses given to patients in clinical trials. No obvious toxicity was noted at the dose used in the present study.
As illustrated in Figures 1 and 2, concurrent use of naproxen or ketoprofen significantly altered the behavior of zalcitabine in rats. Considering that (i) zalcitabine undergoes active tubular secretion in the kidneys[6,7] and that (ii) both zalcitabine and NSAIDs can interact with organic anion transporters in the renal tubular cells[8,9], the reduction of the CLR of zalcitabine in the presence of naproxen or ketoprofen might result, at least in part, from the inhibition of organic anion transporters by naproxen or ketoprofen. Although there are potential adverse effects, these interactions may provide a therapeutic benefit, whereby the interactions prolong the duration of action of zalcitabine, by conferring a longer plasma half-life, thus necessitating less frequent doses of zalcitabine, and also a lower dose. Therefore, the clinical significance of this finding needs to be further evaluated for therapeutic dose levels in clinical studies.
In summary, pretreatment with naproxen or ketoprofen prior to zalcitabine administration significantly altered the pharmacokinetic profile of zalcitabine, implying that patients who are being treated with NSAIDs and zalcitabine may require close monitoring for potential drug interactions.
Acknowledgement
The authors greatly appreciate the help of Mr Ming-ji JIN in carrying out the animal experiments.
References
- Dahlberg JE, Mitsuya H, Blam SB, Broder S, Aaronson SA. Broad spectrum antiretroviral activity of 2',3'-dideoxynucleoides. Proc Natl Acad Sci USA 1987;84:2469-73.
- Ueda K, Tsurimoto T, Nagahata T, Chisaka O, Matsubara K. An in vitro system for screening anti-hepatitis B virus drugs. Virology 1989;169:213-6.
- Waqar MA, Evans MJ, Manly KF, Hughes RG, Huberman JA. Effects of 2',3'-dideoxynucleosides on mammalian cells and viruses. J Cell Phys 1984;121:402-8.
- Barry M, Mulcahy F, Merry C, Gibbons S, Back D. Pharmacokinetics and potential interactions amongst antiretroviral agents used to treat patients with HIV infection. Clin Pharmacokinet 1999;36:289-304.
- Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, et al. A controlled trial of 2 nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N Engl J Med 1997;337:725-33.
- Gustavson LE, Fukuda EK, Rubio FA, Dunton AW. A pilot study of the bioavailability and pharmacokinetics of 2',3'-dideoxycy-tidine in patients with AIDS or AIDS-related complex. J Acquir Immune Defic Synd 1990;3:28-31.
- Klecker RW Jr, Collins JM, Yarchoan RC, Thomas R, McAtee N, Broder S, et al. Pharmacokinetics of 2',3'-dideoxycytidine in patients with AIDS and related disorders. J Clin Pharmacol 1988;28:837-42.
- Gibbs JE, Thomas SA. The distribution of the anti-HIV drug, 2',3'-dideoxycytidine (ddC), across the blood-brain and blood-cerebrospinal fluid barriers and the influence of organic anion transport inhibitors. J Neurochem 2002;80:392-404.
- Wada S, Tsuda M, Sekine T, Cha SH, Kimura M, Kanai Y, et al. Rat multispecific organic anion transporter 1 (rOAT1) transports zidovudine, acyclovir, and other antiviral nucleoside analogs. J Pharmacol Exp Ther 2000;294:844-9.
- Burckhardt BC, Burckhardt G. Transport of organic anions across the basolateral membrane of proximal tubule cells. Rev Physiol Biochem Pharmacol 2003;146:95-158.
- Jariyawat S, Sekine T, Takeda M, Apiwattanakul N, Kanai Y, Sophasan S, et al. The interaction and transport of β-lactam antibiotics with the cloned rat renal organic anion transporter 1. J Pharmacol Exp Ther 1999;290:672-7.
- Khamdang S, Takeda M, Noshiro R, Narikawa S, Enomoto A, Anzai N, et al. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J Pharmacol Exp Ther 2002;303:534-9.
- Uwai Y, Saito H, Hashimoto Y, Inui KI. Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J Pharmacol Exp Ther 2000;295:261-5.
- Ibrahim SS, Boudinot FD. Pharmacokinetics of 2',3'-dideoxycyti-dine in rats: application to interspecies scale-up. J Pharm Pharma-col 1989;41:829-34.
- van Heeswijk RP, Veldkamp AI, Hoetelmans RM, Mulder JW, Schreij G, Hsu A, et al. The steady-state plasma pharmacokinetics of indinavir alone and in combination with a low dose of ritonavir in twice daily dosing regimens in HIV-1-infected individuals. AIDS 1999;13:F95-F99.
- van Praag RM, Weverling GJ, Portegies P, Jurriaans S, Zhou XJ, Turner-Foisy ML, et al. Enhanced penetration of indinavir in cerebrospinal fluid and semen after the addition of low-dose ritonavir. AIDS 2000;14:1187-94.
- Abel S, Nichols DJ, Brearley CJ, Eve MD. Effect of cimetidine and ranitidine on pharmacokinetics and pharmacodynamics of a single dose of dofetilide. Br J Clin Pharmacol 2000;49:64-71.
- Christian CD, Meredith CG, Speeg KV. Cimetidine inhibits renal procainamide clearance. Clin Pharmcol Ther 1984;36:221-7.
- Cundy KC. Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir and adefovir. Clin Pharmacokinet 1999;36:127-43.