
Effects of adenoviral gene transfer of mutated IκBα, a novel inhibitor of NF-κB, on human monocyte-derived dendritic cells1
Introduction
Atopic asthma, characteristic of eosinophilic airway inflammation, is now regarded as a T-helper 2 (Th2) cell-mediated disorder, which is under the control of dendritic cells (DC)[1]. Studies in rodents and humans have revealed the presence of an extensive network of DC in airways[2,3]. Upon maturation in vivo or in vitro, DC become less phagocytic and more potent antigen-presenting cells (APC), expressing high levels of major histocompatibility complex (MHC) class II and costimulatory molecules as well as proinflammatory cytokines. As the most potent professional APC identified, DC play a central role in inducing the primary immune response, priming the naive T cells, initiating the immune tolerance, and regulating the types of T cell responses[4].
Nuclear factor κB (NF-κB), which is typical of p50/p65 heterodimers, is sequestered as inactive trimers in the cytoplasm of quiescent cells through interaction with IκBα, the most important member of the inhibitors of NF-κB (IκB) family. Multiple signals converge on the common signaling pathway for NF-κB activation upon sequential phosphorylation of IκBα at the Ser 32/36 residues of the N-terminal domain, and subsequent proteolytic degradation[5]. Recent data suggest that NF-κB overactivation may be the basis for increased expression of myriad inflammatory genes and for the perpetuation of chronic airway inflammation in asthma[6,7]. More recently, we have cloned the 801 bp Chinese IκBα mutant (IκBαM) gene (203–1003 bp) encoding 267 amino acids from human placenta, a novel nondegradable super-repressor of NF-κB, by site-directed deletion of the N-terminal phosphorylation sites of Ser 32/36, and constructed a replication-deficient recombinant adenovirus vector AdIκBαM[8]. These mutations prevent IκBαM phosphoryla-tion, retaining NF-κB in its inactivated cytosolic location complexed with IκBαM. DC maturation and cytokine production are NF-κB-dependent, thus, we investigated the effects of adenoviral gene transfer of IκBαM on apoptosis, phenotype and function of DC derived from human monocytes to explore a possible approach for future DC-based immunotherapy of asthma.
Materials and methods
Reagents Recombinant human granulocyte/macrophage colony-stimulating factor (GM-CSF), interleukin 4 (IL-4), and tumor necrosis factor α (TNF-α) were generously provided by Dr Guang-yong PENG (Baylor College of Medicine, Dallas, USA). Lipopolysaccharide (LPS) was purchased from Sigma Chemical Co (St Louis, MO, USA). Fluorescein isothio-cyanate (FITC)-conjugated mouse anti-human CD14, HLA-DR, CD83 and CD80 were bought from Serotec (UK), and phycoerythrin (PE)-conjugated mouse anti-human CD86 were bought from Diaclone (USA). Taq DNA polymerase and T4 DNA ligase were obtained from Gibco BRL (USA). [γ-32P] adenosine triphosphate and [3H] thymidine (TdR) were provided by Beijing Furui Biotechnology (China).
Vectors The serotype 5-, E1-, and E3-defective adeno-viruses expressing Escherichia coli β-galactosidase (AdLacZ) and human IκBαM (AdIκBαM) were constructed as describ-ed previously[8]. Remarkably, the molecular weight of novel IκBαM was deduced to be 30 kDa. The generated titers of AdLacZ and AdIκBαM were approximately 1.0×1010 and 4.0×109 pfu/mL, respectively.
Cell culture Blood buffy coat from healthy donors (Jiangsu Institute of Hematology) was centrifuged on Ficoll-Paque (Pharmacia, Sweden) to acquire human peripheral blood mononuclear cells (PBMC). Monocytes were derived from PBMC depleted of NK, B-, and T cells with anti-CD16, anti-CD19 and anti-CD3, as well as goat anti-mouse Ig-conjugated magnetic beads (Miltenyi Biotec, USA) as previously described[9]. These were plated (1×107 cells/3 mL per well) into 6-well plates in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS; Gibco BRL, USA). Cells were cultured at 37 °C in 5% CO2 in medium supplemented with GM-CSF (900 ng/mL) and IL-4 (300 ng/mL). Cultures were fed every 2 d with half of the culture volume of full doses of cytokines. On d 5 cells were stimulated with LPS (100 ng/mL) to induce maturation, and the suspended cells were harvested on d 7.
Electron microscopy On d 7 cells were washed once in phosphate-buffered saline (PBS), resuspended, and fixed overnight in 2% paraformaldehyde and 2.5% glutaraldehyde, followed by fixation for 1 h in 1% cacodylate-buffered osmium tetraoxide. After dehydration in a series of ethanol and propylene oxide solutions, the cells were embedded in epoxy resin, ultrathinly sectioned, stained with uranyl acetate and lead citrate, and examined under a scanning electron microscope (SEM; SX-40; Akashi, Japan). Additionally, the prepared cells were routinely stained and examined under a transmission electron microscope (TEM; JEM-1010; Jeol, Japan).
Gene transfer Adenoviral transfections were performed in 60 mm plates with mature DC at 5×105 cells/plate on d 7. For adenoviral transfections, medium was removed from each plate and replaced with 2 mL of medium containing either AdIκBαM at multiplicity of infection (MOI) of 25, 50, or 100 pfu/cell, or AdLacZ at an MOI of 50 pfu/cell for 4 h, followed by incubation in fresh RPMI-1640 medium supplemented with 10% FCS at 37 °C and in 5% CO2 for an additional 24 h and 48 h, respectively. The untransfected mature DC were used as normal controls.
PCR and RT-PCR DNA and RNA were extracted from mature DC, either untransfected or transfected with AdIκBαM and AdLacZ, using Wizard plasmid isolation and Trizol kits (Promega, USA), according to the manufacturer’s instruc-tions, and the RNA was reverse-transcribed into cDNA for templates. Primers were designed using Primer Express software and were as follows: forward primer 5'-CCTCTAGAA-TGAAAGACGAGGAGTACGAG-3' and reverse primer 5'-TGGTACCTCACAGCTCGTCCTCTGTGAACTCCGTG-3'. The reaction started with an initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at 54 °C for 1 min and extension at 72 °C for 1 min and additional extension of 10 min, after which 5 μL of each product was quantified by 1% agarose gel electro-phoresis.
Western blot analysis After 24 or 48 h of overexpression of the genes of interest, cytosolic proteins were prepared as described elsewhere[10] and quantitated by using the Brad-ford assay. Equivalent amounts of denatured protein (100 µg) were separated on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel, and electrophoretically transferred to nitrocellulose membranes. Nonspecific binding to the membrane was blocked with 5% nonfat dry milk in PBS-Tween overnight at 4 °C. Blots were washed in PBS-Tween, and then probed with anti-human IκBα antibody (1:1000 dilution) and horseradish peroxidase-conjugated anti-goat IgG (1:5000 dilution, Santa Cruz Biotechnology, USA) for 2 h at room temperature. Immunoblots were visualized with an enhanced chemiluminescence detection kit (Amersham Life Sciences, USA).
Electrophoretic mobility shift assay After 48 h over-expression of target genes, DC were harvested upon stimulation with 10 ng/mL TNF-α for 30 min. Preparation of nuclear extracts and the electrophoretic mobility shift assay (EMSA) were carried out as described previously[11]. The NF-κB oligonucleotide (5'-AGTTGAG
Flow cytometry DC that were either untransfected or transfected with AdIκBαM or AdLacZ were incubated at a density of 1×105 cells/100 µL with 10 µL FITC- or PE-conjugated monoclonal antibodies for 30 min at room temperature. The cells were washed once in PBS or fixed with 1% para-formaldehyde, then the surface markers of mature DC were assessed by FACScan (Becton Dickinson, USA). In addition, apoptosis of mature DC was immediately analyzed by FACScan after staining with 5 µL annexin V (AV)-FITC and 5 µL propidium iodide (PI) for 20 min at 4 °C. The total cell population consisted of DC that were AV single positive (apoptotic), PI single positive (necrotic), AV and PI double positive (apo-necrotic), or AV and PI double negative (live).
Enzyme-linked immunosorbent assay Supernatants from mature DC were harvested 48 h after transfection with AdIκBαM and AdLacZ. Levels of IL-12 (p70) were determined using a sandwich enzyme-linked immunosorbent assay (ELISA) kit (PharMingen, USA) in compliance with the manufacturer’s instructions.
Mixed leukocyte reaction Allogeneic T cells were obtained from nylon wool nonadherent PBMC by negative selection, with a purity of more than 95%[12]. T cells were cultured in triplicate in 96-well flat-bottomed plates at 2×105 cells/well, to which nonautologous mature DC, either untrans-fected or transfected with AdIκBαM and AdLacZ, were added at 2×103, 4×103, 10×103, or 20×103 cells/well. Plates were incubated at 37 °C and 5% CO2 for 96 h, and pulsed with 0.5 µCi/well of [3H]thymidine for the final 16 h before harvesting on filters for scintillation counting. Counts per minute (cpm) of triplicate wells were deduced by subtracting the background cpm of the medium alone.
Statistical analysis Data were expressed as mean±SD, and assessed by one-way analysis of variance (ANOVA) and paired Student’s t-tests with SPSS 11.0 software. Statistical difference was assumed at P<0.05.
Results
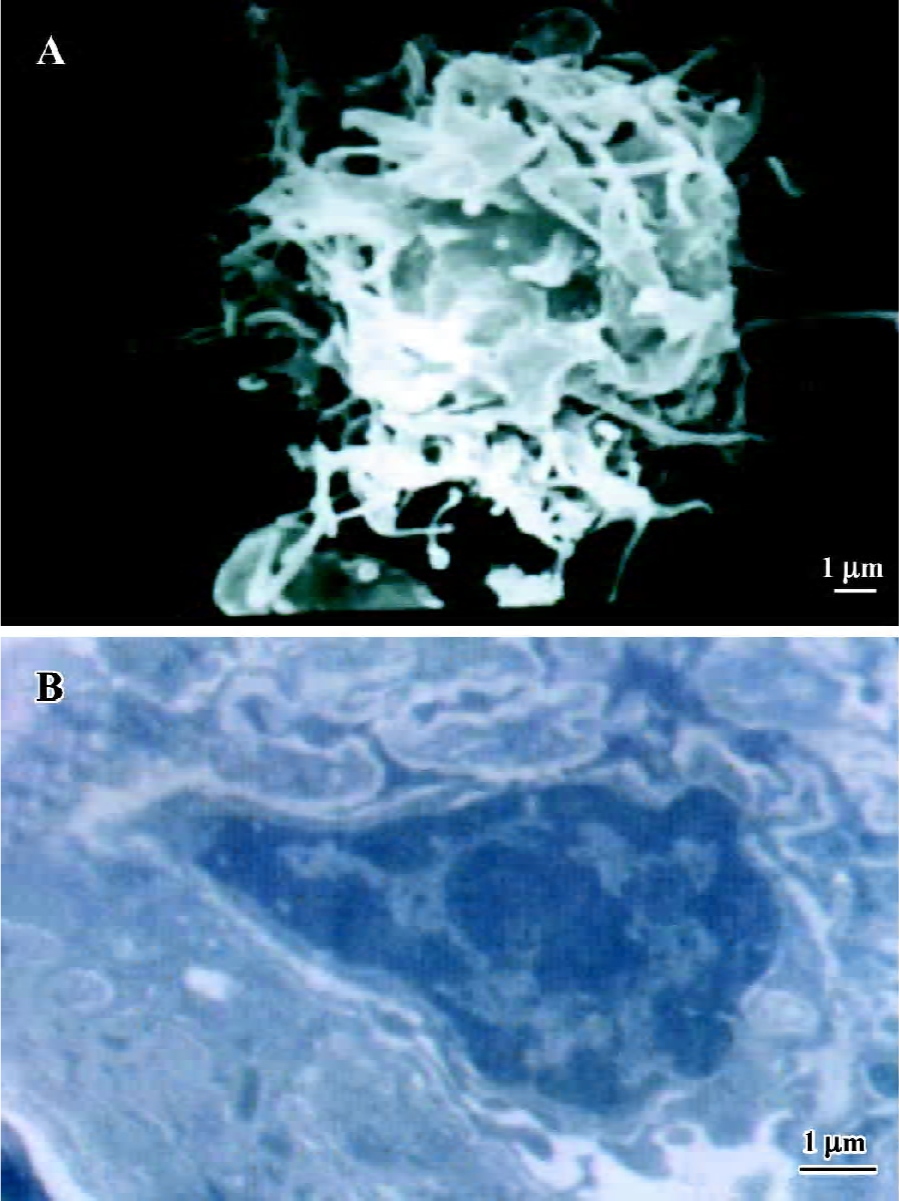
Morphology of mature DC In culture with GM-CSF and IL-4 for 5 d, monocytes differentiated into mature DC after stimulation with LPS for 2 d. The mature DC showed prominent dendritic and veiled projections (>10 µm) on d 7 under the SEM (Figure 1A), and displayed plentiful surface dendrites and cytosolic vesicles mostly of endocytic type, but a relatively immature Golgi zone under the TEM (Figure 1B).
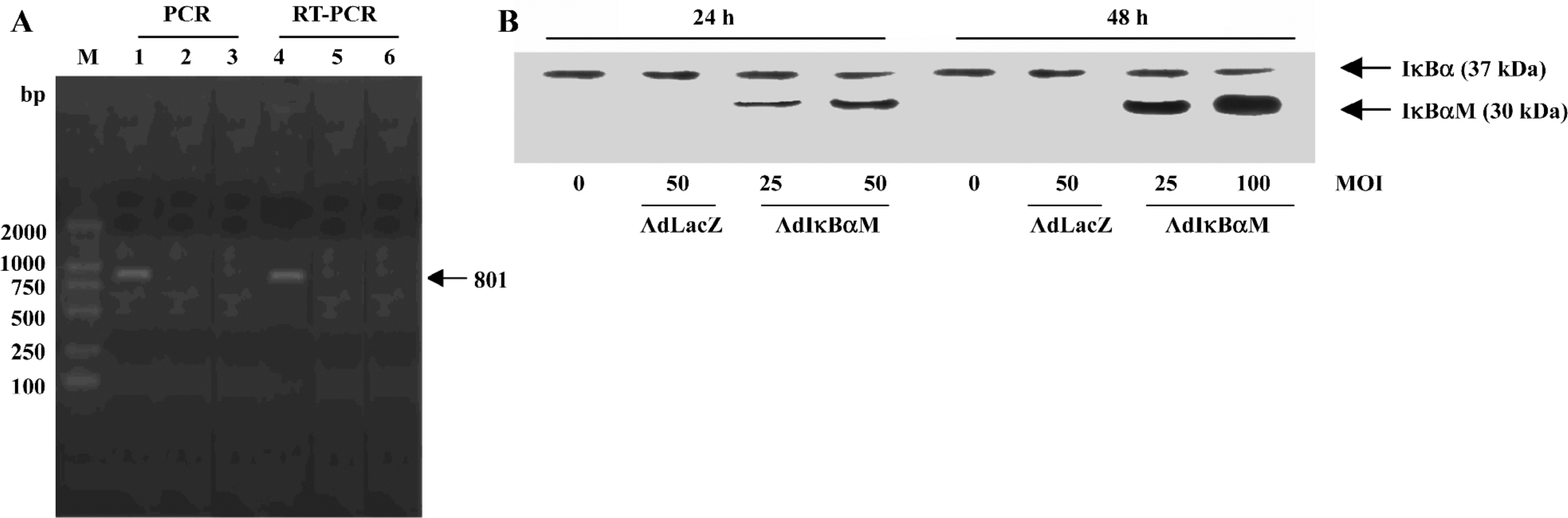
Detection of IκBαM gene in mature DC Electrophoretic analysis of both PCR and RT-PCR products revealed the unique 801 bp IκBαM cDNA in AdIκBαM-transfected, but not in AdLacZ-transfected or untransfected, mature DC, indicating that the IκBαM gene is integrated into the genome and transcribed in infected mature DC (Figure 2A). As determined by Western blot analysis, the tagged transgene product can be visualized as being slightly larger than the native IκBα. A dose- and time-dependent increase in IκBαM expression can be seen in AdIκBαM-transfected mature DC, which peaked at an MOI of 100 pfu/cell and at 48 h (Figure 2B).
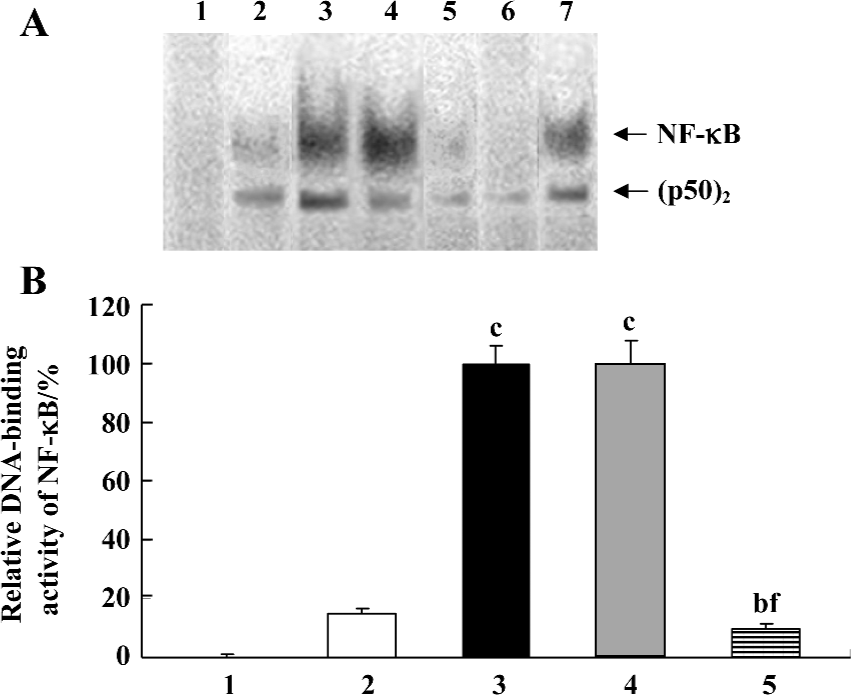
Inhibition by AdIκBαM of TNF-α-induced NF-κB activation in mature DC The function of AdIκBαM in blocking NF-κB binding was assessed by EMSA. In contrast to a basal NF-κB activity in the control (untransfected and nonstimulated) DC, the TNF-α-stimulated DC exhibited a dramatically promoted NF-κB activity (P<0.01). Gene transfer of IκBαM, but not LacZ, at an MOI of 100 pfu/cell and 48 h sharply inhibited the NF-κB activity compared with the TNF-α-stimulated DC (P<0.01), suggesting that AdIκBαM inhibits TNF-α-induced NF-κB activation in mature DC (Figure 3A, 3B). In competition assays, addition of 100-fold excess of unlabeled NF-κB, but not mutant NF-κB, oligonucleotide affected NF-κB binding, confirming the specificity of NF-κB binding (Figure 3A).
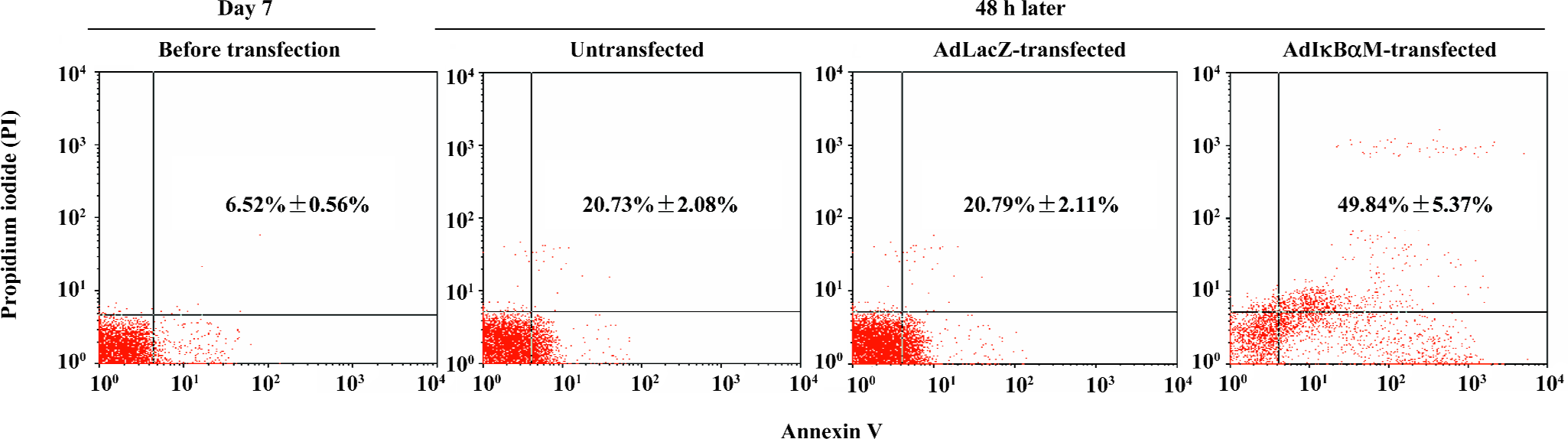
Induction by AdIκBαM of apoptosis in mature DC Flow cytometry revealed low levels of normal apoptosis in mature DC on d 7, with a higher level of apoptosis 48 h later (P<0.01). Optimal gene transfer of IκBαM, but not LacZ, significantly augmented the apoptosis of mature DC (P<0.01), demonstrating that AdIκBαM facilitates apoptosis in mature DC (Figure 4).
Effects of AdIκBαM on phenotype and function of mature DC Flow cytometry revealed low levels of CD14, but high levels of HLA-DR, CD83, CD80, and CD86 in mature DC. Except for similar levels of CD14 and HLA-DR (P>0.05), optimal gene transfer of IκBαM, but not LacZ, greatly downregulated the levels of CD83, CD80, and CD86 in mature DC, among which CD80 had a smaller decrease than did CD86 (all P<0.01). Notably, the mature DC overexpressing IκBαM still secreted high levels of CD83, which is a hallmark of maturation. These results imply that downregulation of the B7 surface molecules by AdIκBαM in mature DC may contribute to the immune shift of T cells (Figure 5, Table 1). ELISA confirmed that gene transfer of IκBαM, but not LacZ, resulted in a lower secretion levels of IL-12 (p70) in mature DC (P<0.01, Figure 6). The mixed leukocyte reaction revealed, in parallel with the ratio of DC:T cells, a similar potency for stimulating the proliferation of allogeneic T cells between AdLacZ-transfected and untransfected mature DC. In contrast, the AdIκBαM-transfected mature DC embodied a strikingly decreased mixed leukocyte reaction in comparison with the AdLacZ-transfected mature DC (P<0.05 or 0.01; Figure 7).
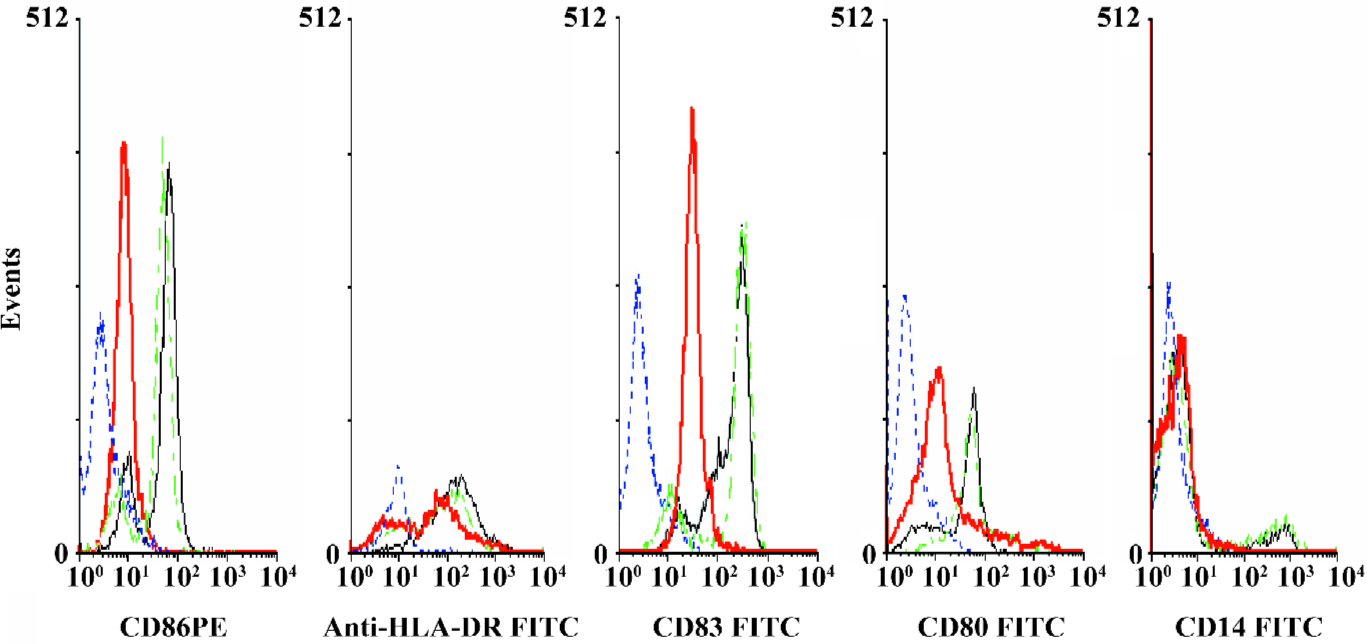
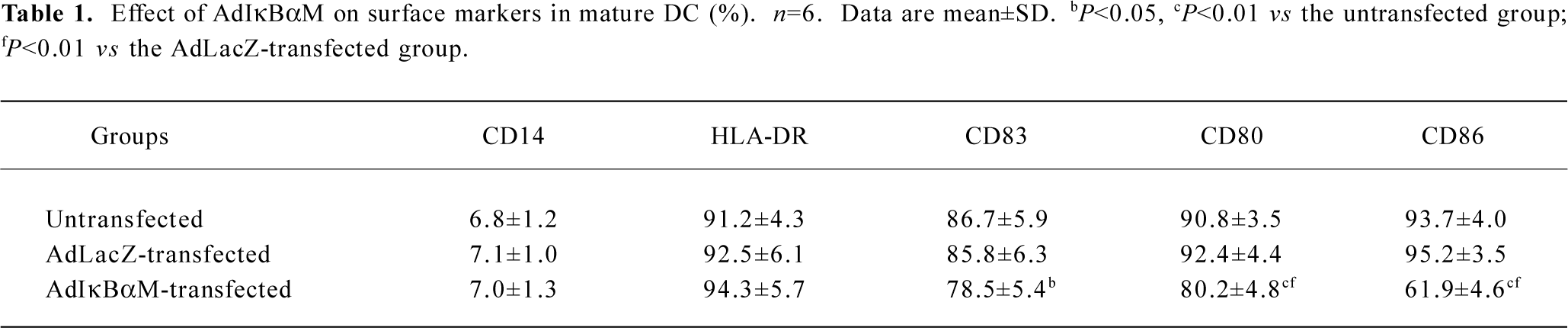
Full table
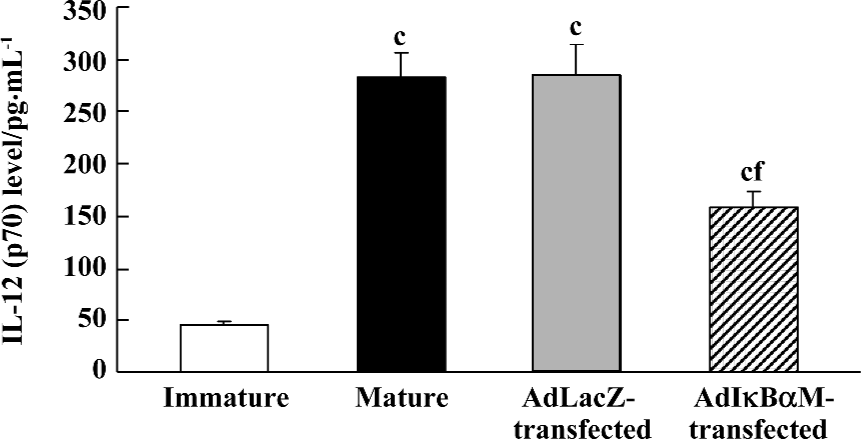
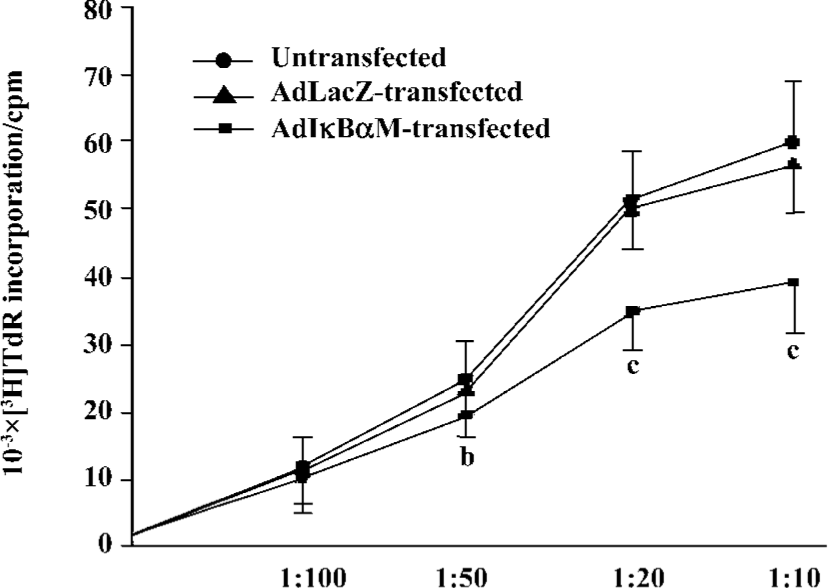
Discussion
T-cell stimulation and Th1/Th2-cell polarization require 3 DC-derived signals: signal 1 (recognizing signal), which is mediated through T-cell receptor (TCR) triggering by MHC class II-associated peptides processed from pathogens; signal 2 (costimulatory signal), which is mainly mediated by triggering of T cell CD28 by CD80 (B7-1) and CD86 (B7-2); and signal 3 (polarizing signal), which is mediated by various soluble or membrane-bound factors[13–15].
NF-κB plays a crucial role in antiapoptosis by regulation of the transcription of cytokine genes, induction of the antiapoptotic genes, and promotion of the TNF receptor-associated factors and the inhibitor-of-apoptosis proteins to block the activation of caspase-8[16,17]. IκBα, a 37 kDa inhibitory protein, is arranged in 3 domains: the N-terminal domain-containing specific phosphorylation sites, the internal domain of 5 tandem ankyrin repeat sequences, and the C-terminal domain containing Pro-Glu-Ser-Thr polypeptides for regulation of NF-κB activity, binding of NF-κB, and rapid protein turnover, respectively[18]. IκBα retains the p65-containing NF-κB complex in the cytoplasm by masking its nuclear localization sequence (NLS). Upon appropriate stimulation, for example by IL-1β, TNF-α, or LPS, IκBα undergoes phosphorylation on two serine residues, Ser 32 and Ser 36, which renders IκBα susceptible to proteolytic degradation via the ubiquitin-proteasome pathway, resulting in release and nuclear translocation of NF-κB. Consequently, the active NF-κB, single or in combination with other nuclear factors, initiates transcription of target genes associated with immune and inflammatory responses. As for the inflammatory cascade, NF-κB repression in airways through suppression of IκBα degradation or augmentation of IκBα synthesis can downregulate transcription of an array of the NF-κB-dependent genes, which is more effective than blockade of single downstream inflammatory or immune genes[19].
In the present study, monocytes cocultured with GM-CSF and IL-4 differentiated into mature DC upon stimulation with maturation inducer LPS. The IκBαM gene was successfully delivered into the monocyte-derived mature DC, and was expressed time- and dose-dependently. Importantly, NF-κB-blocked mature DC by AdIκBαM obviously inhibited the proliferation of T cells, which is in agreement with the report that selective inhibition of NF-κB in DC by the NF-κB essential modulator-binding domain peptide blocks maturation and prevents T-cell proliferation, accompanied by less Th1 and Th2 polarization[20]. Furthermore, TNF-α-induced NF-κB activation of mature DC was significantly attenuated by AdIκBαM, and the NF-κB-blocked mature DC were more susceptible to apoptosis than those transfected with AdLacZ. Nevertheless, blockade of NF-κB in mature DC, other than little change in HLA-DR related to antigen-presentation, caused striking downregulation of the B7 costimulatory molecules. It is deduced, therefore, that AdIκBαM might, in part, facilitate the antigen-specific immune tolerance of T cells via induction of DC apoptosis and downregulation of B7 molecules.
Asthma is, per se, a multigenetically susceptible clinical syndrome, in which sensitized individuals develop an aberrant Th2-dominated immunity for the underlying basis of eosinophilic airway inflammation[10,14]. B7-1 and B7-2 differentiate the Th0 cells into a Th1 subset for cellular immunity and a Th2 subset for humoral immunity, respectively[1,13,15]. Interestingly, downregulation of B7-2 was more effective than that of B7-1 in the mature DC overexpressing IκBαM, indicating that blockade of NF-κB in DC may, to some extent, contribute to rectifying the Th1/Th2 imbalance in asthmatics. It is noteworthy that Th2 development could be the default pathway induced by DC when the production of IL-12 is quite low[2]. As the major effective treatment for asthma, corticosteroids, like all pharmacologic agents that have been shown to inhibit NF-κB, have numerous other effects that could limit their therapeutic usefulness[21]. Currently, there is much interest in identifying more specific and effective NF-κB inhibitors, among which the IκBαM is a novel NF-κB inhibitor. Although blocking the NF-κB pathway is unlikely to be a clinically beneficial approach due to the broad range of genes involved, it is of importance to understand the role of NF-κB during specific immune responses for identifying relevant molecules as potential therapeutic targets[22].
In summary, our findings suggest that the AdIκBαM is applicable to both effective gene transfer of IκBαM and specific blockade of NF-κB in monocyte-derived mature DC. Moreover, the ability of mature DC overexpressing IκBαM to induce DC apoptosis and downregulate B7 molecules highlights an important mechanism for T-cell immune suppression. The successful downregulation of T-cell responses following inhibition of NF-κB in mature DC offers a potential strategy for future immunotherapy of asthma[23,24]. Antigen-specific T-cell immune tolerance could be induced by selective blockade of NF-κB with AdIκBαM in antigen-primed mature DC (ie by pollen, ragweed, house dust mite, etc). Alternatively, the DC-mediated Th1-dominated response is promising for amelioration of the undue Th2 by AdIκBαM in combination with Th1-inducing agents, such as CpG oligodeoxynucleotide, purified protein derivative and bacilli Calmette-Guerin[25,26].
Acknowledgements
We would like to thank Prof Joel N KLINE (Division of Pulmonary Medicine, University of Iowa Hospitals and Clinics) and Prof Gang HU (Department of Pharmacology and Neurobiology, Nanjing Medical University) for helpful comments and critical review of the manuscript.
References
- Zhou LF, Yin KS. Toll-like receptors: function and roles in asthma. Chin Med J 2004;117:1709-15.
- Mazzoni A, Young HA, Spitzer JH, Visintin A, Segal DM. Histamine regulates cytokine production in maturing dendritic cells, resulting in altered T-cell polarization. J Clin Invest 2001;108:1865-73.
- Zhou LF, Yin KS, Zhou ZM. Role of low dosage arsenic trioxide on pulmonary dendritic cells in asthmatic mice. Chin J Integr Med 2003;9:281-4.
- Blanco P, Palucka KA, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science 2001;294:1540-3.
- Christman JW, Sadikot RT, Blackwell TS. The role of nuclear factor κB in pulmonary diseases. Chest 2000;117:1482-7.
- Woodruff PG, Fahy JV. Asthma: prevalence, pathogenesis, and prospects for novel therapies. JAMA 2001;286:395-8.
- Zhou LF, Yin KS, Zhu ZL, Yao X, Xie WP, Mao H, et al. Inhibition of nuclear factor κB activation through induction of IκBα expression by arsenic trioxide in a murine model of asthma. Respirology 2004;9 Suppl:A94.
- Zhou LF, Yin KS, Zhu ZL, Yi Zhu, Yao X, Mao H, et al. Adenovirus-mediated overexpression of novel mutated IκBα inhibits nuclear factor κB activation in endothelial cells. Chin Med J 2005;118:1422-8.
- Palucka KA, Taquet N, Sanchez-Chapuis F, Gluckman JC. Dendritic cells as the terminal stage of monocyte differentiation. J Immunol 1998;160:4587-95.
- Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, et al. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science 2004;304:1678-82.
- Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T-cell-mediated suppression by dendritic cells. Science 2003;299:1033-6.
- Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, Toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med 2002;196:1645-51.
- Lambrecht BN. The dendritic cell in allergic airway diseases: a new player to the game. Clin Exp Allergy 2001;31:206-18.
- Zhou LF, Yin KS. The role of B7-CD28/CTLA-4 costimulatory pathway in bronchial asthma. Chin J Cell Mol Immunol 2002;18:656-8.
- Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol 2003;3:984-93.
- Cui XF, Imaizumi T, Yoshida H, Tanji K, Matsumiya T, Satoh K. Lipopolysaccharide induces the expression of cellular inhibitor of apoptosis protein-2 in human macrophages. Biochim Biophys Acta 2000;1524:178-82.
- Fang F, Wang AP, Yang SF. Antitumor activity of a novel recombinant mutant human tumor necrosis factor-related apoptosis-inducing ligand. Acta Pharmacol Sin 2005;26:1373-81.
- Zhao GH, Vaszar LT, Qiu DM, Shi LF, Kao PN. Anti-inflammtory effects of triptolide in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 2000;279:L958-66.
- Fan C, Li Q, Ross D, Engelhardt JF. Tyrosine phosphorylation of I kappa B alpha activates NF-kappa B through redox-regulated and c-Src-dependent mechanism following hypoxia/reoxygenation. J Biol Chem 2003;278:2072-80.
- Tas SW, de Jong EC, Hajji N, May MJ, Ghosh S, Vervoordeldonk MJ, et al. Selective inhibition of NF-κB in dendritic cells by the NEMO-binding domain peptide blocks maturation and prevents T-cell proliferation and polarization. Eur J Pharmacol 2005;35:1164-74.
- Jamaluddin M, Choudhary S, Wang S, Casola A, Huda R, Garofalo RP, et al. Respiratory syncytial virus-inducible BCL-3 expression antagonizes the STAT/IRF and NF-kappa B signaling pathways by inducing histone deacetylase 1 recruitment to the interleukin-8 promoter. J Virol 2005;79:15302-13.
- Calder VL, Bondeson J, Brennan FM, Foxwell BMJ, Feldmann M. Antigen-specific T-cell downregulation by human dendritic cells following blockade of NF-κB. Scand J Immunol 2003;57:261-70.
- Luft C, Hausding M, Finotto S. Regulation of T-cells in asthma: implications for genetic manipulation. Curr Opin Allergy Clin Immunol 2004;4:69-74.
- Xie WP, Wang H, Ding JH, Wang H, Hu G. Anti-proliferating effect of iptakalim, a novel KATP channel opener, in cultured rabbit pulmonary arterial smooth muscle cells. Eur J Pharmacol 2005;511:81-7.
- Ji Y, Han Y, Diao JX, Huang Y, Chen Q, Ferro A. Rabbit aortic endothelial dysfunction by low-density lipoprotein is attenuated by L-arginine, L-ascorbate and pyridoxine. Br J Pharmacol 2003;140:1272-82.
- Peng GY, Guo Z, Kiniwa Y, Voo KS, Peng WY, Fu TH, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T-cell function. Science 2005;309:1380-4.