
Role of 11-beta-hydroxysteroid dehydrogenase type 1 in differentiation of 3T3-L1 cells and in rats with diet-induced obesity1
Introduction
Obesity is now reaching epidemic proportions in some countries, and has significant health implications. Lifestyle advice alone rarely results in long-term weight reduction. Thus, the pharmacological targeting of metabolic pathways (ie insulin resistance, dyslipidemia, and hypertension) in obese patients may be the best option to help prevent obesity-related cardiovascular disease[1].
11-β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1), as a potential therapeutic target in obesity and the associated “metabolic syndrome”, is an attractive research focus. 11β-HSD1 knockout mice have low intracellular glucocorticoid levels, and are protected from obesity, diabetes, and dyslipidemia[2,3]. Conversely, transgenic overexpression of 11β-HSD1 in white adipose tissue results in mice with elevated intracellular glucocorticoid levels, central obesity, insulin resistance, hypertension, hyperglycemia, and dyslipidemia[4–6]. Recently, studies have also shown that a marked downregulation of 11β-HSD1 activity occurs with high fat (HF) feeding in mice[7] and that there is no change in 11β-HSD1 with HF feeding in Wistar rats[8]. The reason for this difference remains unclear, but similar interindividual variations in 11β-HSD1 may contribute to metabolic disease.
Metabolic syndrome comprises a variety of disorders, including type 2 diabetes and hyperlipidemia, which relate to the loss of insulin sensitivity in important target tissues such as adipose tissue, muscle and liver[9,10]. 11β-HSD1 catalyzes an in vivo conversion of inactive to active glucocorticoids. But the mechanisms by which elevated body fat causes reduced insulin sensitivity, and the roles of 11β-HSD1 and glucocorticoids in obesity are poorly understood.
The 3T3-L1 preadipocyte model and a well-characterized outbred rat obesity model are widely utilized in obesity studies[11]. We used the in vitro model in the current study. The aim of our study was to explore the role of 11β-HSD1 in 3T3-L1 cells differentiation and observe the gene expression of 11β-HSD1 in diet-induced obesity rats. Thus, the association of obesity and insulin resistance could be provoked by regionally altered steroid responsiveness with 11β-HSD1 playing a central role.
Materials and methods
Culture and differentiation of 3T3-L1 cells 3T3-L1 preadipocytes were cultured and induced to differentiate as described previously[12]. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), in 5% CO2. Two days after the cells were confluent (d 0), they were induced to differentiate by changing the medium to DMEM containing 10% FBS, plus 0.5 mmol/L 3-isobutyl-1-methyxanthine (MIX; Sigma, St Louis, MO, USA), 5 mg/mL insulin (Sigma), and 1 mmol/L dexamethasone (Sigma). After 48 h (d 2), the medium was replaced with DMEM containing 10% FBS, plus 5 mg/mL insulin, and the medium was changed every 2 d for 8 d.
Oil red O staining Cells were stained with oil red O as described previously[13]. Briefly, 3T3-L1 cells were fixed in 10% formalin (pH 7.4) for 30 min, and then 3T3-L1 cells were stained with oil red O for 5 min (stock solution: 3 mg/mL dissolved in isopropanol; working solution: 60% oil red O stock stain combined with 40% distilled water).
Animal and diets Thirty male Sprague-Dawley (SD) rats (90–120 g Animal Center of Jiangsu Province) were housed at 21–23 °C with light provided from 7:00 to 19:00. All rats were allowed free access to food and water for 6 d, then they were divided into 2 groups. One group of rats (diet-induced obesity; DIO) were switched to a high-energy (HE) diet of 8% coin oil, 44% sweetened condensed milk, and 48% rat chow, and another group (controls) were switched to rat chow (Xietong Animal Feed Factory, Nanjing, China). Bodyweight, total length, and tail length (tip of tail to anus) were recorded at 9:00 every day, and body length was calculated (total length minus tail length).
After 2 weeks, the HE diet groups had increased weight and Lee’s index (obesity value index; weight1/3×103/body length). They received the HE diet for 6 more weeks, and the controls continued to be fed rat chow. The experiments were conducted in accordance with a specific institutions guidelines.
Laboratory examination of blood samples Blood glucose, serum insulin, triglyceride (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and TNF-α levels were measured. Samples of heart blood were collected and serum was separated for assay. Glucose, cholesterol and TG levels were measured by using enzymatic methods (Boehringer-Mannheim German) adapted for an automatic analyzer (Hitachi 717). HDL-C and LDL-C concentrations were obtained by using the same method. Insulin and TNF-α were analyzed by radioimmunoassay (Linco).
Western blot analysis Total protein from the 3T3-L1 cells and animal tissues was suspended in a RIPA buffer, containing 150 mmol/L NaCl, 50 mmol/L Tris-HCl, 1% Triton X-100, 5 mmol/L deoxycholic acid sodium salt, 0.2% sodium dodecylsulfate (SDS) with 1× Halt protease inhibitor cocktail (Pierce) mixed using a ultra Turrax T25 homogenizer (IKA Laboratechnik). Lysates were incubated on ice for 20 min, then centrifuged at 13 000×g for 15 min at 4 °C. Ten milligrams of protein were subjected to 12% SDS-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride (PVDF) membranes, and the membrane was blocked in Tris-buffered saline containing 15% powder milk and 0.1% Tween-20. Immunoblotting was carried out with 11β-HSD1 rabbit anti-human primary antibody (at 4 °C overnight; Santa Cruz). β-actin antibody was used for internal controls (Sigma). After incubation with the primary antibody, membranes were washed with 1× phosphate-buffered saline (PBS) containing 0.1% Tween-20 4 times. Secondary antibodies, goat anti-rabbit IgG-horseradish peroxidase conjugate (Santa Cruz) were used, respectively, for 11β-HSD1 and β-actin antibodies (at room temperature for 2 h). After washing, equal amounts of enhanced chemiluminescent reagent (ECL; Amersham Pharmacia Biotech) were applied to membranes for 1 min. Membranes were then wrapped in plastic, placed in X-ray film cassettes containing Kodak Medical X-ray film (Kodak Medical Systems), and exposed in a dark room for 45–120 s. The exposed film was scanned using a densitometer (Furi, Shanghai) to determine the optical densities of each band, and the density ratio of 11β-HSD1 to β-actin bands was calculated. All experiments were repeated for triplicate.
RNA preparation and amplification by real-time polymerase chain reaction Real-time reverse-transcription polymerase chain reaction (RT-PCR) quantitative analysis of mRNA was applied for 11β-HSD1 and GR and some marker genes of preadipocyte differentiation. Total RNA was isolated from 3T3-L1 cells by using the Trizol method (Invitrogen). One microgram of total RNA was converted to cDNA using 200 U Moloney murine leukemia virus reverse transcriptase (Promega, USA) in a volume of 20 μL, which contained 0.5 mmol/L dNTP, 20 U RNase inhibitor, and 0.5 μg oligo (deoxythymidine) 15 primer (Promega). A control experiment without reverse transcriptase was performed for each sample in order to verify that the amplification did not come from genomic DNA contamination. PCR primers were designed by Primer Designer software based on sequences downloaded from GenBank and were custom-made by Invitrogen Biotechnology, as shown in Table 1. Each PCR was carried out in triplicate in a 20 μL volume using QuantiTect SYBR Green PCR Master Mix (MJ Research) for 10 min at 94 °C after initial denaturing for 5 min, followed by 44 cycles of 94 °C for 20 s, 58 °C for 30 s and 72 °C for 30 s in an ABI Prism 7700 sequence detection system. Values for each gene were normalized against the expression levels of β-actin bands was calculated. All experiments were repeated for triplicate.
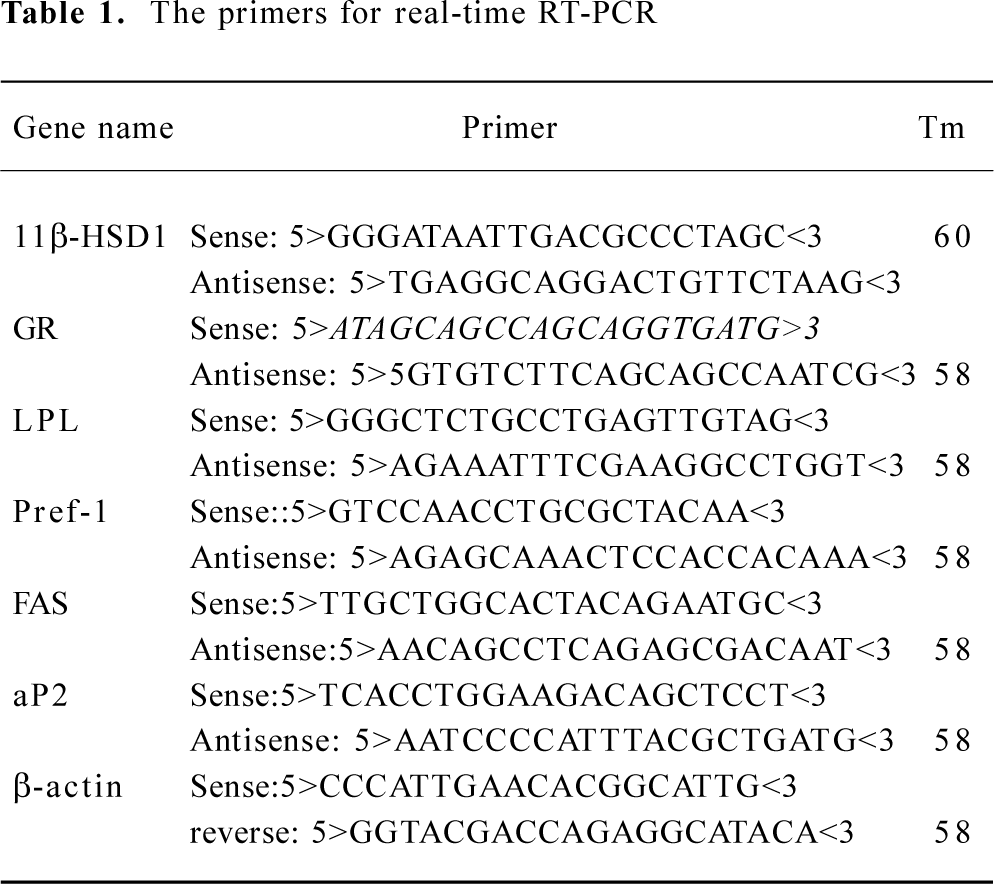
Full table
Statistical analysis All results are expressed as mean±SD. Data were analyzed using one-way ANOVA with a correction for multiple comparisons, as appropriate.
Results
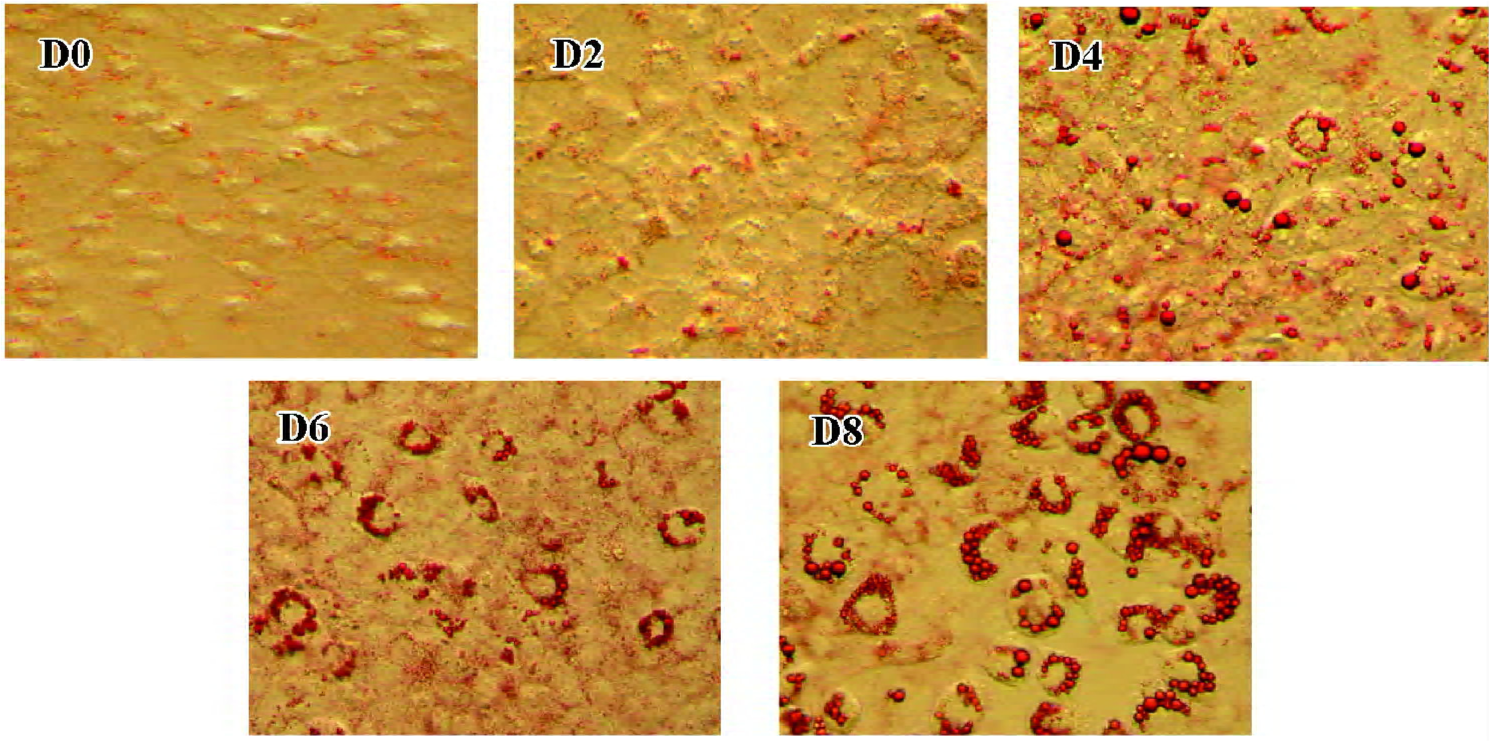
3T3-L1 preadipocyte model On d 0, 2, 4, 6 and 8 of 3T3-L1 cell differentiation, oil red O staining was used to detect lipid droplets. Lipid droplets accumulated after stimulation (d 0), and the degree of lipid droplet accumulation increased with differentiation (Figure 1; the amplified multiples were ×20).
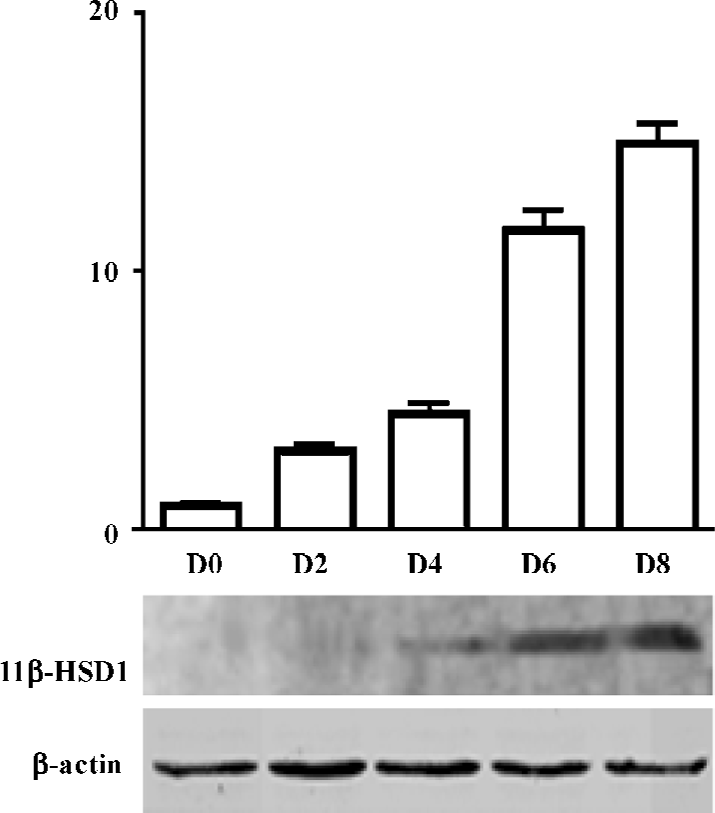
Expression of 11β-HSD1 protein, during adipocyte differentiation (d 0, 2, 4, 6, 8), was detected by Western blot analysis. 11β-HSD1 protein was detected as a major band at 34 kDa, and was confirmed to be present in 3T3-L1 cells. Expression of 3T3-L1 protein was upregulated, and accompanied the process of adipocyte differentiation (d 8 vs d 0, 11β-HSD1 relative value was 295.67±25.54 vs 11.00± 0.01; P<0.01; Figure 2).
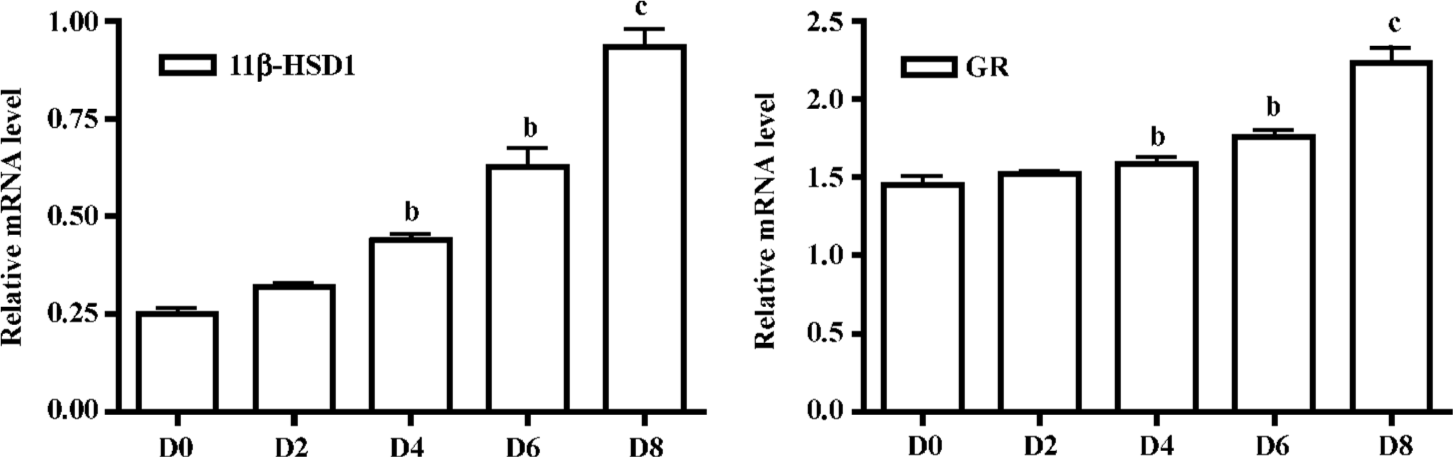
Expression of 11β-HSD1 and GR mRNA were detected by real-time RT-PCR analysis on d 0, 2, 4, 6 and 8. mRNA levels were upregulated after incubation on d 4 and especially in mature adipocytes (Figure 3).
The mRNA expression pattern of some 3T3-L1 cell differentiation markers [eg LPL, Pref-1, aP2, and fatty acid synthetase (FAS)] were also detected at the same time. Early upregulation in LPL expression was detected in our study (d 4 vs d 0, 3.77±0.21 vs 2.01±0.20, P<0.05; Figure 4). This has often been cited as an early sign of adipocyte differentiation, and important transcription factors related to adipogenesis were induced early in adipocyte differentiation. Abundant expression of Pref-1 was detected in pre-adipocytes. How-ever, a dramatic decrease in expression that occurred with adipocyte differentiation was found, and lower levels were detected in mature fat cells (d 8 vs d 0, 1.02±0.06 vs 1.68±0.01, P<0.01; Figure 4).
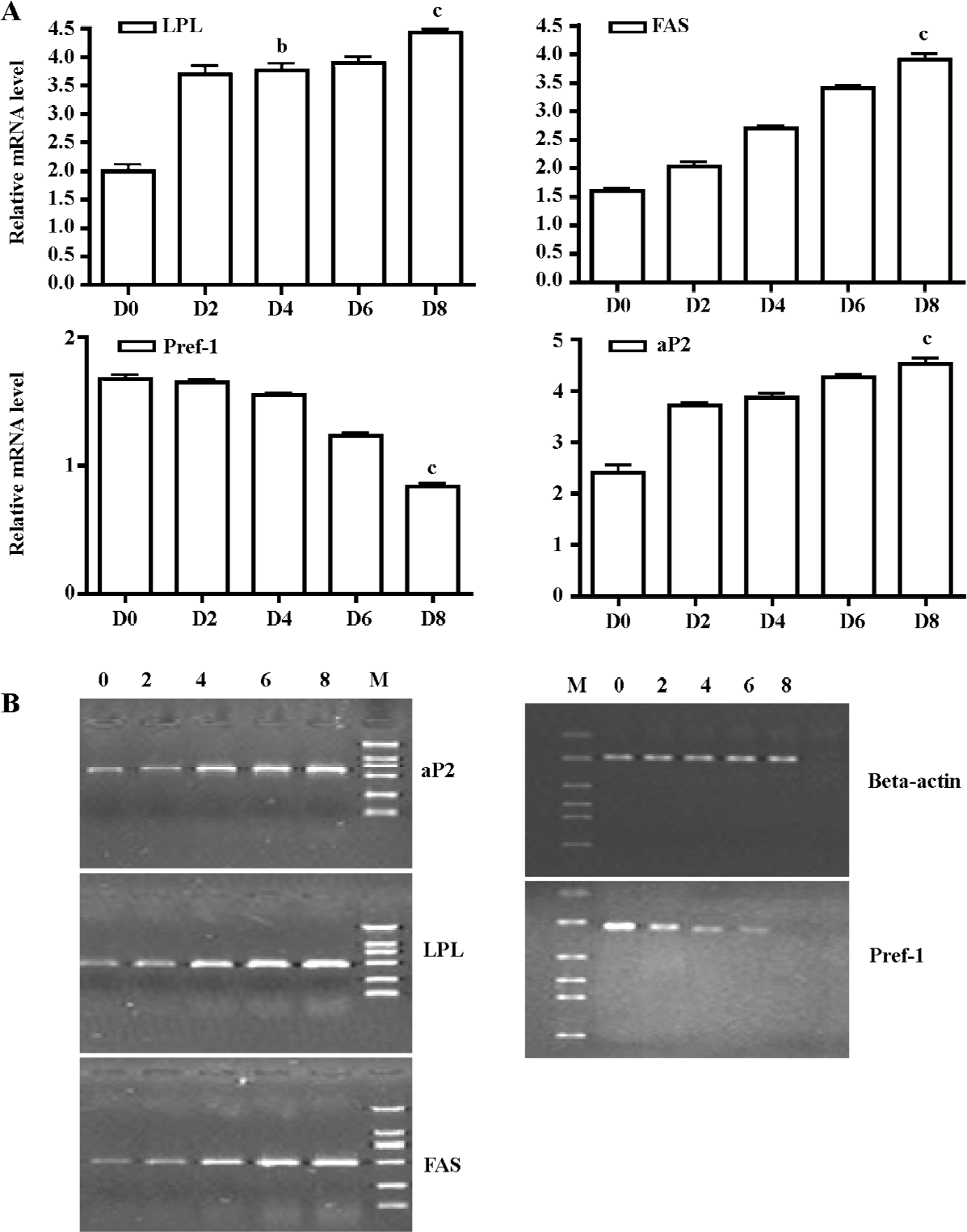
Some markers of a late stage of adipocyte differentiation, such as aP2 (d 8 vs d 0, 4.48±0.01 vs 2.33±0.02, P<0.01) and FAS (d 8 vs d 0, 3.90±0.20 vs 1.60±0.10, P<0.01; Figure 4) were also detected. Our results showed that most markers had an expression pattern that involved later upregulation, and were in accordance with the expression of 11β-HSD1 and GR.
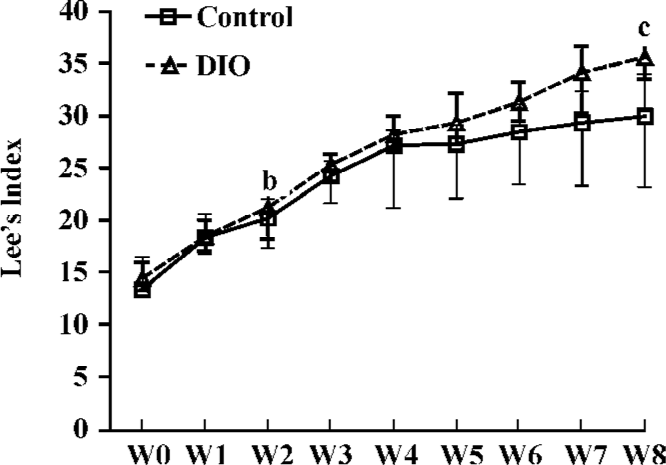
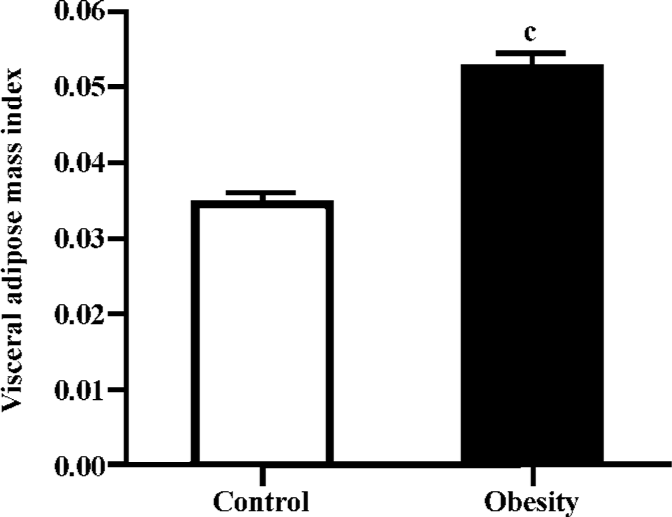
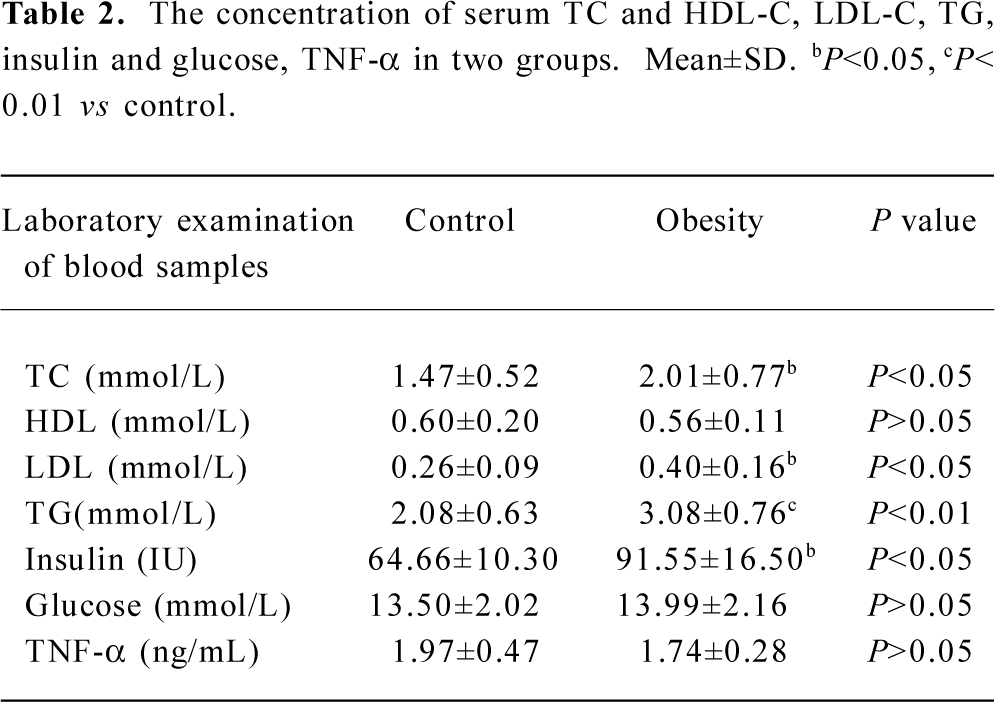
DIO rat model As shown in Figure 5, after 2 weeks on the HE diet, the DIO rats had significantly greater body weight and Lee’s index values than the control rats (P<0.05). Visceral adipose mass showed the same trends as body-weight, and the weight of visceral adipose mass was higher in DIO rats than in control rats (P<0.01) (Figure 6). The serum concentrations of TC (P<0.05), LDL (P<0.05), and TG (P<0.01) were significantly different, but there was no difference in HDL-C between the DIO and control rats. There was significantly increased insulin concentrations in DIO rats, but there was no difference in blood glucose (P>0.05) between the DIO and control rats. The concentration of serum TNF-α was not different in DIO rats and controls (Table 2).
Full table
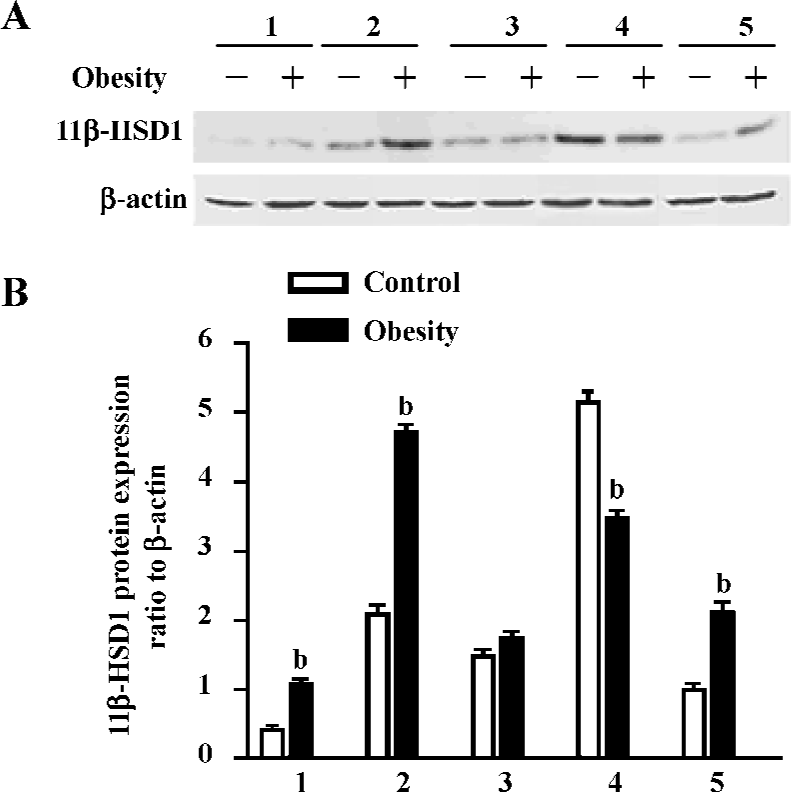
To better understand the role of 11β-HSD1 in the development of obesity and insulin resistance, the protein expression of 11β-HSD1 in tissues of the 2 groups were detected by Western blotting, and the results showed that the protein expression of 11β-HSD1 was significantly upregulated in visceral adipose, brain and muscle tissue of the DIO rats, but that it was downregulated in the liver (Figure 7).
Discussion
11β-HSD1 predominantly acts as an oxoreductase that forms active glucocorticoids from inactive keto-glucocor-ticoids, and because it is associated with metabolic syndrome, it is a potential therapeutic target in obesity. However, the role of glucocorticoids in obesity is poorly understood[1]. 3T3-L1 preadipocytes and rats with DIO, have been widely used in studies on obesity. We used these two models to explore the relationship between 11β-HSD1 and preadipocyte differentiation, and the role of 11β-HSD1 in obesity.
Oil red O staining results and real time RT-PCR analyses of adipogenesis marker genes indicated that the 3T3-L1 model was appropriate for the study of preadipocyte differentiation (Figures 1, 4).
Lee’s index, the weight of visceral adipose mass (Figures 5, 6), and the serum TG and TC results (Table 2) indicated that the DIO mice represented an obesity model that was appropriate for our study of 11β-HSD1 and obesity. Glucocorticoids play an important role in normal physiology by modulating metabolic and immune responses[14]. At a cellular level, their actions are mediated via inhibition of cell proliferation and induction of differentiation[15]. There is accumulating evidence suggesting that glucocorticoids play a role in adipocyte maturation. Cortisol contributes to the process that inhibits cellular proliferation, but strongly promotes adipocyte differentiation. In general, glucocorticoids inhibit cellular proliferation by inducing cell cycle arrest at the G1 phase; the pre-receptor modulation of cortisol metabolism has a dramatic effect on the cellular proliferation rate[14,15]. Along with other steroid hormones, glucocorticoids act by binding to cognate intracellular GR[16,17]. The resulting nuclear complex acts as a ligand-dependent trans-activator, either by binding as a homodimer to specific target gene response elements, or by protein-protein interactions with other transcriptional regulators[18,19]. Although it is now clear that glucocorticoids regulate the transcription of a diverse array of target genes, the precise mechanisms by which GR-mediated changes in cell proliferation and differentiation occur are still far from clear[15]. Data presented here emphasize the pivotal role of 11β-HSD1 as a pre-receptor determinant of GR-mediated signal transduction. A recent study shows that adipocytes use 11β-HSD1 to regulate their differentiation in an autocrine manner[20], which supplies adequate amounts of cortisol to support cell differentiation and prevents the accumulation of excessive and detrimental amounts of cortisol in mature cells. The present study reveals the importance of autocrine 11β-HSD1 action for proper cell differentiation and function. Cortisol induces adipogenesis and 11β-HSD1 activity in preadipocytes. When adipocyte differentiation is initiated, 11β-HSD1 dehydrogenase activity is switched into reductase activity, generating cortisol and thereby promoting adipocyte differentiation[14,21,22].
In adipose stromal cells, 11β-HSD1 has also been shown to regulate the glucocorticoid induction of aromatase activity. Our data has indicated the importance of 11β-HSD1 expression in 3T3-L1 cells. 11β-HSD1 protein and mRNA were significantly upregulated during the process of preadipocyte differentiation into mature adipocytes, and reached a maximum 6–8 d after confluence. At the same time the GR level was also upregulated during 3T3-L1 cell differentiation (Figure 3). Our data support the hypothesis that 11β-HSD1-mediated reductase activity and expression increase with differentiation in 3T3-L1 mouse cells, and that 11β-HSD1, as a stimulator of preadipocyte differentiation, may serve as a maker gene in obesity[11,15]. Further studies are needed to explore the regulation and functional pathways of 11β-HSD1 at the gene and protein levels. Glucocorticoids also play a fundamental role in the differentiation and growth of animal tissues by modulating gene expression. They exert their effect through binding to intracellular receptors (GR) with subsequent stimulation or repression of target gene trans-cription.
The last decade has witnessed an exponential increase in research focusing on 11β-HSD1, principally because of its putative role in human obesity and insulin resistance[23–26]. 11β-HSD1 knockout mice have low intracellular glucocorticoid levels and are protected from obesity, diabetes, and dyslipidemia[2,3]. Conversely, transgenic overexpression of 11β-HSD1 in white adipose tissue produces mice with elevated intracellular glucocorticoids and central obesity, insulin resistance, hypertension, hyperglycemia, and dyslipidemia[5,6].
The 11β-HSD1 knockout mouse does not display fasting hypoglycemia in the basal state; however, when fed with a HE diet, fasting glucose levels are significantly lower than in wild type controls[27]. Transgenic mice overexpressing hepatic 11β-HSD1 appear to have elevated insulin levels following a glucose load, as well as dyslipidemia and hypertension, but more detailed data with respect to hepatic glucose flux in these animals and the molecular mechanism of 11β-HSD1 and obesity are as yet not available.
Our study showed that 11β-HSD1 activity in DIO rat liver was decreased (Figure 7), but there was no difference in glucose levels between DIO rats and controls, although insulin levels were increased in DIO rats (Table 2). These findings do not agree with those of Rohner-Jeanrenaud, who found that adipose tissue 11β-HSD1 mRNA expression was increased at the very onset of HE diet-induced obesity in mice (ie at d 2), and that this increase was positively correlated with the degree of hyperglycemia[28].
In one study, a marked downregulation of activity was found with HE feeding in mice [7]. This may represent a short-term adaptive mechanism to limit the well-established adverse metabolic consequences of HE feeding[8,29]. However, unlike in mice, with chronic HE feeding, Wistar rats exhibit a relative loss of the potentially protective HE-mediated downregulation of adipose 11β-HSD1, as shown in the present study (Figure 7). The reason for this difference remains unclear, although the failure to maintain reduced adipose and/or hepatic 11β-HSD1 activity may be important in the pathogenesis of the metabolic sequelae associated with obesity and, indeed, may mean that Wistar rats are more susceptible than mice to the metabolic consequences of dietary obesity. These data support the hypothesis that variation in susceptibility to obesity and its metabolic consequences may, in part, be caused by interindividual differences in susceptibility to the dysregulation of 11β-HSD1[7]. Furthermore, they suggest a novel and complementary hypothesis that a similar interindividual variation in hepatic clearance/metabolism of glucocorticoids by 5α-reductase may also contribute to metabolic disease susceptibility.
11β-HSD1 is also expressed in skeletal muscle. The role of tissue-specific cortisol metabolism within muscle and its impact upon insulin sensitivity has not been extensively studied. However, levels of expression within human skeletal myoblasts have been found to correlate with measures of insulin resistance, Lee’s index and blood pressure [30]. The characterization of polymorphisms within the human 11β-HSD1 gene has enabled an evaluation of this locus as a sensitive factor for obesity. Our study also demonstrated 11β-HSD1 activity in rat muscle, and revealed that expression was higher in DIO rats than in controls (Figure 7). There-fore, inhibitors of 11β-HSD1 for treatment of obesity and diabetes mellitus should be not only selective for adipose tissues and liver, but also be effective in muscle.
High levels of NADP-dependent 11β-HSD1 dehydrogenase activity have also been detected in rat brain; however, the sensitivity of the brain to glucocorticoids is not only determined by GR, but also by 11β-HSD1[31]. Studies have also supported the hypothesis that 11β-HSD1 in the brain is acting primarily as a reductase[32,33], which has been supposed to be important in explaining their improved age-related learning impairments in comparison with controls. Additionally, in the hypothalamus/pituitary, inhibition of 11β-HSD1 with glycyrrhetinic acid has been shown to modulate the negative glucocorticoid feedback mechanism by inhibiting corticotrophin releasing factor (CRF) concentrations within hypophysial portal blood[34], and also altered cerebral glucose metabolism[14], all of which suggests functional dehydrogenase activity at these sites. Our results demonstrated that there was 11β-HSD1 activity in rat brain, and that the expression of 11β-HSD1 in the brain was higher in DIO than in control rats, and that it was expressed at higher levels in the brain than in muscles. There was no significant difference in the concentration of blood glucose between the two groups, although TC, LDL and TG were higher in DIO than in control rats. It is proposed that the expression of 11β-HSD1 in the brain may play an important role in obesity by reverse feedback. However, further research is needed to explain the molecular mechanisms and the pathway of function of 11β-HSD1 at the gene and protein levels.
Numerous studies have been directed toward understanding the effects of various steroid moieties upon 11β-HSD1 activity, given that any factors that inhibit metabolism of the 11β-hydroxyl group will increase glucocorticoid potency. Several studies have now demonstrated that increased 11β-HSD1 activity in adipose tissue is associated with changes in mRNA expression, suggesting that transcriptional regulation increases the expression of glucocorticoid-sensitive target genes such as resistin, CAATT enhancer binding proteins (C/EBP), peroxisome proliferator activiated receptor (PPAR-γ) and adipocyte factor TNF-α. Although there has been extensive study of glucocorticoid metabolism in monogenic forms of obesity in rodents, little is known about the effects of dietary obesity on glucocorticoids[28]. In our 3T3-L1 cell differentiation model, the expression of these marker genes was correlated with the expression of 11β-HSD1 and GR (data not shown). But TNF-α levels were not different in DIO rats or in controls (Table 2). It can be hypothesized that these regulators may partially act via local stimulation of 11β-HSD1, which would favor adipose tissue glucocorticoid action signaling in diet-induced obesity models. A study indicates the presence of a molecular switch that regulates 11β-HSD1 promoter activity depending on the state of cellular differentiation[22], but further study is certainly needed.
Currently, newer selective 11β-HSD1 inhibitors are in development. The present study suggested that inhibition of 11β-HSD1 could be a successful therapy in obesity and diabetes mellitus. Elucidation of the complex mechanisms of 11β-HSD1 in obesity would certainly be a significant achievement.
References
- Wake DJ, Walker BR. 11beta-Hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Mol Cell Endocrinol 2004;215:45-54.
- Kotelevtsev YV, Holmes MC, Burchell A. 11beta-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid inducible responses and resist hyperglycaemia on obesity or stress. Proc Natl Acad Sci USA 1997;94:14924-9.
- Morton NM, Holmes MC, Fievet C. Improved lipid, lipoprotein profile, hepatic insulin sensitivity, and glucosetolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem 2001;276:41293-300.
- Walker BR. beta-Hydroxysteroid dehydrogenase type 1 in obesity. Obes Res 2004;12:1-3.
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl J R, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001;294:2166-70.
- Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, et al. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest 2003;112:83-90.
- Nicholas MM, Lyner R, Jonathan RS. Down-regulation of adipose 11beta-hydroxysteroid dehydrogenase type 1 by high-fat feeding in mice: a potential adaptive mechanism counteracting metabolic disease. Endocrinology 2004;145:2707-12.
- Drake AJ, Livingstone DE, Andrew R, Seckl JR, Morton NM, Walker BR. Reduced adipose glucocorticoid reactivation and increased hepatic glucocorticoid clearance as an early adaptation to high-fat feeding in wistar rats. Endocrinology 2005;146:913-9.
- Reaven GM, Hoffman BB. A role for insulin in the aetiology and course of hypertension? Lancet 1987;2:435-7.
- Waldhäusl WK, Roden MD. The effects of free fatty acids on glucose transport and phosphorylation in human skeletal muscle. Curr Opin Endocrinol Metabol 2000;7:211-6.
- Felmer RN, Clark JA. The gene suicide system ntr/cb1954 causes ablation of differentiated 3t3l1 adipocytes by apoptosis. Biol Res 2004;37:449-60.
- Blagoev B, Kratchmarova I, Nielsen MM, Fernandez M M, Voldby J, Andersen J S, et al. Inhibition of adipocyte differentiation by resistin-like molecule alpha. Biochemical characterization of its oligomeric nature. J Biol Chem 2002;277:42011-6.
- Sahai A, Patel MS, Zavosh AS, Tannen RL. Chronic hypoxia impairs the differentiation of 3t3-l1 fibroblast in culture: role of sustained protein kinase c activation. J Cell Physiol 1994;160:107-12.
- Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper M S, et al. 11β-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 2004;25:831-66.
- Rabbitt EH, Lavery GG, Walker EA, Cooper MS, Stewart PM, Hewison M. Prereceptor regulation of glucocorticoid action by 11beta-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J 2002;16:36-44.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835-9.
- Reichardt HM, Schutz G. Glucocorticoid signalling: multiple variations of a common theme. Mol Cell Endocrinol 1998;146:1-6.
- Konig H, Ponta H, Rahmsdorf HJ, Herrlich P. Interference between pathway-specific transcription factors: glucocorticoids antagonize phorbol ester-induced ap-1 activity without altering ap-1 site occupation in vivo. EMBO J 1992;11:2241-6.
- Wei P, Inamdar N, Vedeckis WV. Transrepression of c-jun gene expression by the glucocorticoid receptor requires both ap-1 sites in the c-jun promoter. Mol Endocrinol 1998;12:1322-33.
- Eijken M, Hewison M, Cooper MS, de Jong FH, Chiba H, Stewart PM, et al. 11beta-Hydroxysteroid dehydrogenase expression and glucocorticoid synthesis are directed by a molecular switch during osteoblast differentiation. Mol Endocrinol 2005;19:621-31.
- Bujalska IJ, Walker EA, Hewison M, Stewart PM. A switch in dehydrogenase to reductase activity of 11 beta-hydroxysteroid dehydrogenase type 1 upon differentiation of human omental adipose stromal cells. J Clin Endocrinol Metab 2002;87:1205-10.
- Bujalska IJ, Kumar S, Hewison M, Stewart PM. Differentiation of adipose stromal cells: the roles of glucocorticoids and 11beta-hydroxysteroid dehydrogenase. Endocrinology 1999;140:3188-96.
- Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM, Arlt W, et al. Mutations in the genes encoding 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet 2003;34:434-9.
- Lindsay RS, Wake DJ, Nair S, Bunt J, Livingstone DE, Permana PA, et al. Subcutaneous adipose 11beta-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in pima Indians and Caucasians. J Clin Endocrinol Metab 2003;88:2738-44.
- Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Heintze U, Janke J, et al. Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res 2004;12:9-17.
- Draper N, Echeald SM, Lavery GG. Association studies between microsatellite markers within the gene encoding human 11beta-hydroxysteroid dehydrogenase type 1 and body mass index, waist to hip ratio, and glucocorticoid metabolism. J Clin Endocrinol Metab 2002;87:4984-90.
- Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA 1997;94:14924-9.
- Asensio C, Muzzin P, Rohner-Jeanrenaud F. Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int J Obes Relat Metab Disord 2004;28 Suppl 4:S45-52.
- DeFronzo RA, Ferrannini E. Insulin resistance: A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991;14:173-94.
- Whorwood CB, Donovan SJ, Flanagan D, Phillips DI, Byrne CD. Increased glucocorticoid receptor expression in human skeletal muscle cells may contribute to the pathogenesis of the metabolic syndrome. Diabetes 2002;51:1066-75.
- Velkoska E, Cole TJ, Morris MJ. Early dietary intervention: long-term effects on blood pressure, brain neuropeptide Y, and adiposity markers. Am J Physiol Endocrinol Metab 2005;288:E1236-43.
- Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1-a tissue-specific amplifier of glucocorticoid action. Endocrinology 2001;142:1371-6.
- Yau JL, Noble J, Kenyon CJ, Hibberd C, Kotelevtsev Y, Mullins JJ, et al. Lack of tissue glucocorticoid reactivation in 11beta-hydroxysteroid dehydrogenase type 1 knockout mice ameliorates age-related learning impairments. Proc Natl Acad Sci USA 2001;98:4716-21.
- Seckl JR, Dow RC, Low SC, Edwards CR, Fink G. The 11beta-hydroxysteroid dehydrogenase inhibitor glycyrrhetinic acid affects corticosteroid feedback regulation of hypothalamic corticotrophin-releasing peptides in rats. J Endocrinol 1993;136:471-7.