
Prostaglandin A1 inhibits increases in intracellular calcium concentration, TXA2 production and platelet activation1
Introduction
Prostaglandins are derived from arachidonic acid liberated from the cell membrane. They are produced in a variety of tissues and mediate an array of physiologic and pathologic processes, including regulation of respiratory, vascular, intestinal and inflammatory activities[1]. Cyclopentenone prostaglandins (cycPG), including the A and J series, are formed from the enzyme-induced dehydration of prostaglandin E and D. They have some unique biological activities that are different from those of classic prostaglandins, such as inducing cell differentiation[2], anti-tumor[3–5] and anti-virus activities[6,7]. Prostaglandin A1 (PGA1) has many pharmacological actions, including inhibiting tumor growth, inflammation and viral replication[8−11]. We have reported previously that PGA1 inhibits excitotoxin-induced apoptosis of striatal neurons in vivo and rotenone-induced apoptosis of cultured SH-SY5Y cells[12,13]. In a recent study, we found that PGA1 significantly reduced infarction volume in rodent models of focal cerebral ischemia[14,15]. PGA1 influences several cellular signaling pathways, including activation of peroxisome proliferator-activated receptor γ (PPARγ)[16], inhibition of nuclear factor-kappaB (NF-κB)[11,17], and induction of heat shock proteins[18,19]. The molecular signaling pathways involved in its neuroprotection, however, remain to be determined.
The activation and aggregation of platelets play an important role in the pathological process of cerebral ischemia through interactions with endothelial cells and formation of thrombosis[20,21]. It has been shown that drugs inhibiting platelet function, including aspirin, could be beneficial in preventing ischemic attack. The effects of some prostaglandins on platelets have been reported[22,23]. However, information about the effects of PGA1 on platelets is incomplete and inconsistent; therefore, the aim of the present study was to determine if PGA1 had an inhibitory effect on the functions of human platelets. We found that PGA1 inhibited platelet aggregation, release of TXB2 and platelet adhesion to endothelial cells. These actions could contribute to its neuroprotective effects in rodent models of stroke.
Materials and methods
Drugs and reagents PGA1, thrombin, Fluo-3 acetoxymethyl ester (Fluo-3 AM), sulfinpyrazone, and A23187 were purchased from Sigma (St Louis, MO, USA); adenosine 5' diphosphate (ADP), N-(2-hydroxyethylpiperazine)-NV-(2-ethanesulfonic acid) (HEPES), and ethylene glycol bi-β-aminoethylether)-N,N,NV-tetraacetic acid (EGTA) were purchased from Shanghai Sangon Bioengineering and Technology Service (Shanghai, China). Collagen was purchased from the Jiangsu Institute of Hematology (Suzhou, China).
Platelet preparations Venous blood was obtained from healthy donors with no drug history for at least 14 d, and immediately mixed with a one-ninth volume of citrate acid (3.8%). Platelet enriched plasma (PRP) was obtained by centrifugation at 200×g for 10 min. For experiments with thrombin, platelets were washed with HEPES-buffed solution (140 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 10 mmol/L glucose and 20 mmol/L HEPES), and then resuspended in Tyrode’s solution (137 mmol/L NaCl, 2.7 µmol/L KCl, 0.36 µmol/L NaH2PO4, 12 µmol/L NaHCO3, 1 µmol/L MgCl2, 5 µmol/L HEPES, 5 µmol/L glucose, 2 µmol/L CaCl2).
Platelet aggregation assay Aggregation of platelets was evaluated by using an MPG-3E aggregometer (Shanghai Silong Medical Instrument Factory, Shanghai, China) according to the manufacturer’s instructions. PRP was first incubated with ethanol (with a final concentration of 0.8%, as a vehicle control), or different concentrations of PGA1 at 37 °C for 5 min. Platelet aggregation was triggered by adding 20 µL of ADP, collagen and thrombin to 200 µL of PRP to achieve final concentrations of 40 µmol/L, 40 µg/mL and 0.25 U/mL, respectively.
Platelet adhesion assay Platelets were washed and treated as described earlier. Human umbilical endothelial cells were cultured in 96-well plates and grown to cover the entire surface of wells. Endothelial cells were pre-treated with Triton X-100 (0.5%) before use. Thrombin-stimulated platelet suspension was added to each well and incubated for 30 min. The adherent platelets were stained with Rose Bengal (0.25%) at 37 °C and then dissolved in absolute ethanol 30 min later. The number of adherent platelets was determined by measuring absorption using a wavelength of 570 nm.
Assay of 5-hydroxytryptamine release from platelets Levels of 5-hydroxytryptamine (5-HT) released from platelets were determined by using O-phthaldialdehyde (OPT) fluorospectrophotometry as described elsewhere[2]. Briefly, platelets were obtained and washed as described earlier, and different concentrations of PGA1 (20–80 µmol/L) or vehicle (final concentration=0.8% ethanol) were incubated with washed human platelets for 5 min. 5-HT was extracted after stimulation with thrombin (0.25 µmol/L) for 5 min and reacted with OPT in 10 mol/L HCl for 15 min. Fluorescence was then measured with a 960 CRT fluorospectrophotometer (Shanghai Jinmi Scientific Instrument Company, Shanghai, China) using excitation and emission wavelengths of 365 nm and 480 nm, respectively. The concentration of 5-HT was calculated as follows: [5-HT]=(Fsample–Fblank)/(Fstandard–Fblank)×0.5 (µg/mL)
Evaluation of ultrastructure of platelets The effect of PGA1 on ultrastructural changes of platelets induced by thrombin was examined with an electron microscope. Washed platelets were pretreated with PGA1 (20–80 µmol/L), then thrombin (0.25 U/mL) was added to activate platelets 5 min later. Platelets were then fixed with 4% (w/v) glutaraldehyde after a 5 min incubation with thrombin, stained with osmium tetroxide, dehydrated, and embedded in Araldite. Ultrathin sections were examined with a Philips CM-120 electron microscope.
TXB2 assay Platelets were obtained as described earlier. Washed platelets were prepared and pre-incubated with PGA1 (20–80 µmol/L) or vehicle (final concentration=0.8% ethanol) for 5 min. Then thrombin was added and the reaction was terminated (500 µL of ice-cooled stop solution containing 50 mmol/L EDTA; 2 mmol/L indomethacin, 130 mmol/L NaCl) 5 min later. Samples were then centrifuged at 500×g for 10 min at 4 °C. Supernatants were used for determination of the levels of TXB2 with radioimmunoassay (TXB2 Radioimmunoassay kit, Jiangsu Institute of Hematology).
Determination of cytosolic free calcium The intracellular [Ca2+] was determined using Fluo-3 AM, essentially as described elsewhere[1]. Briefly, PRP was incubated with 8 µmol/L Fluo-3 AM for 30 min at 37 °C; the dyed platelets were spun down and gently re-suspended at a concentration of approximately 1×108 cells/mL in the HEPES-buffered solution containing 140 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 10 mmol/L glucose and 20 mmol/L HEPES, supplemented with 100 mmol/L sulfinpyrazone to prevent the cellular efflux of Fluo-3 acid. The external [Ca2+] was adjusted to 1 mmol/L and the fluorescence was measured with flow cytometer using excitation and emission wavelengths of 488 nm and 526 nm, respectively. Calibration of the ratio of fluorescence signals into pseudo fluorescence was performed using 1 µmol/L A23187 to obtain the maximal ratio, followed by 5 mmol/L EGTA to obtain the minimal ratio. Platelets were incubated with PGA1 (20–80 µmol/L) at 37 °C for 5 min in HEPES-buffered solutions before the addition of thrombin. The full response was obtained in solutions containing 1 mmol/L CaCl2. Calcium concentration was estimated as follows: Fp=(F–Fmin)/(Fmax–Fmax). To clarify whether the rise in intracellular calcium is derived from intracellular reservoirs or the entry of extracellular Ca2+, platelets were stimulated in a Ca2+-free solution, and calcium concentration was estimated as described earlier.
Statistical analysis Statistical analyses of the differences between vehicle control and PGA1-treated samples were carried out with an unpaired, two-tailed Student’s t-test, and P<0.05 was considered significant.
Results
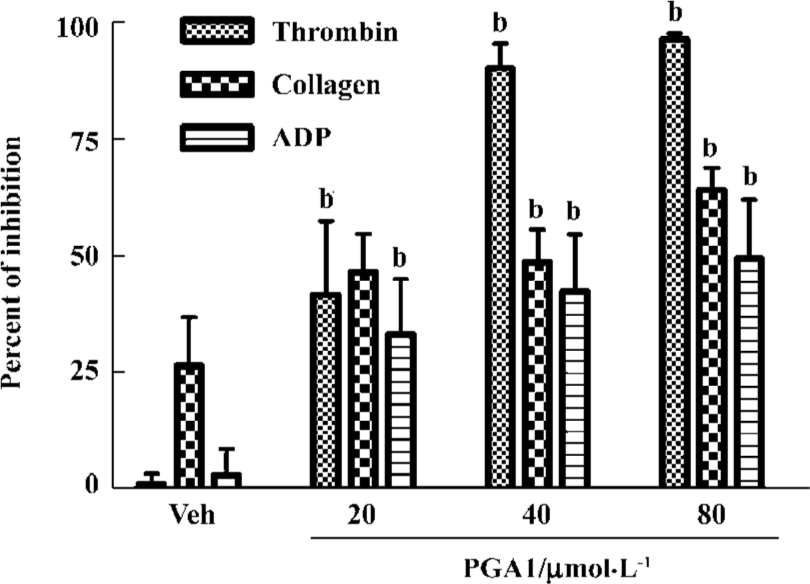
PGA1 inhibited platelet activation Adding ADP (40 μmol/L), collagen (40 mg/mL) or thrombin (0.25 U/mL) to human platelets triggered robust aggregation. Pretreatment with PGA1 (20–80 µmol/L) dose-dependently inhibited the platelet aggregation induced by collagen, ADP and thrombin. The maximal inhibition of PGA1 (20, 40, and 80 µmol/L) on platelet aggregation induced by collagen was 46.63%, 48.70%, and 64.11% (P<0.05); that by ADP was 33.13%, 42.45%, and 49.43% (P<0.05); and that by thrombin was 41.63%, 90.29%, and 96.39% (P<0.05), respectively (Figure 1). The IC50 values of PGA1 on platelet aggregation induced by collagen, ADP and thrombin were 31.02, 80.56, and 20.67 μmol/L, respectively. Thus, PGA1 had greater effects on the thrombin-induced aggregation of platelets. Thrombin-induced adhesion of platelets to endothelial cells was inhibited by pretreatment with PGA1 5 min before thrombin. The inhibitory rates of PGA1 at concentrations of 20 µmol/L, 40 µmol/L, and 80 µmol/L on platelet adhesion were 38.7%, 34.1%, and 40.8%, respectively (P<0.05 vs vehicle; Figure 2). PGA1 significantly decreased the release of 5-HT from the dense granules of platelets induced by thrombin (P<0.05; Figure 3). The lactate dehydrogenase (LDH) assay showed that there was no difference in LDH leakage among groups with and without PGA1 treatment using the same samples for 5-HT assay, excluding the possibility that the increase in 5-HT in supernatant was caused by damage of platelets during preparation (data not shown).
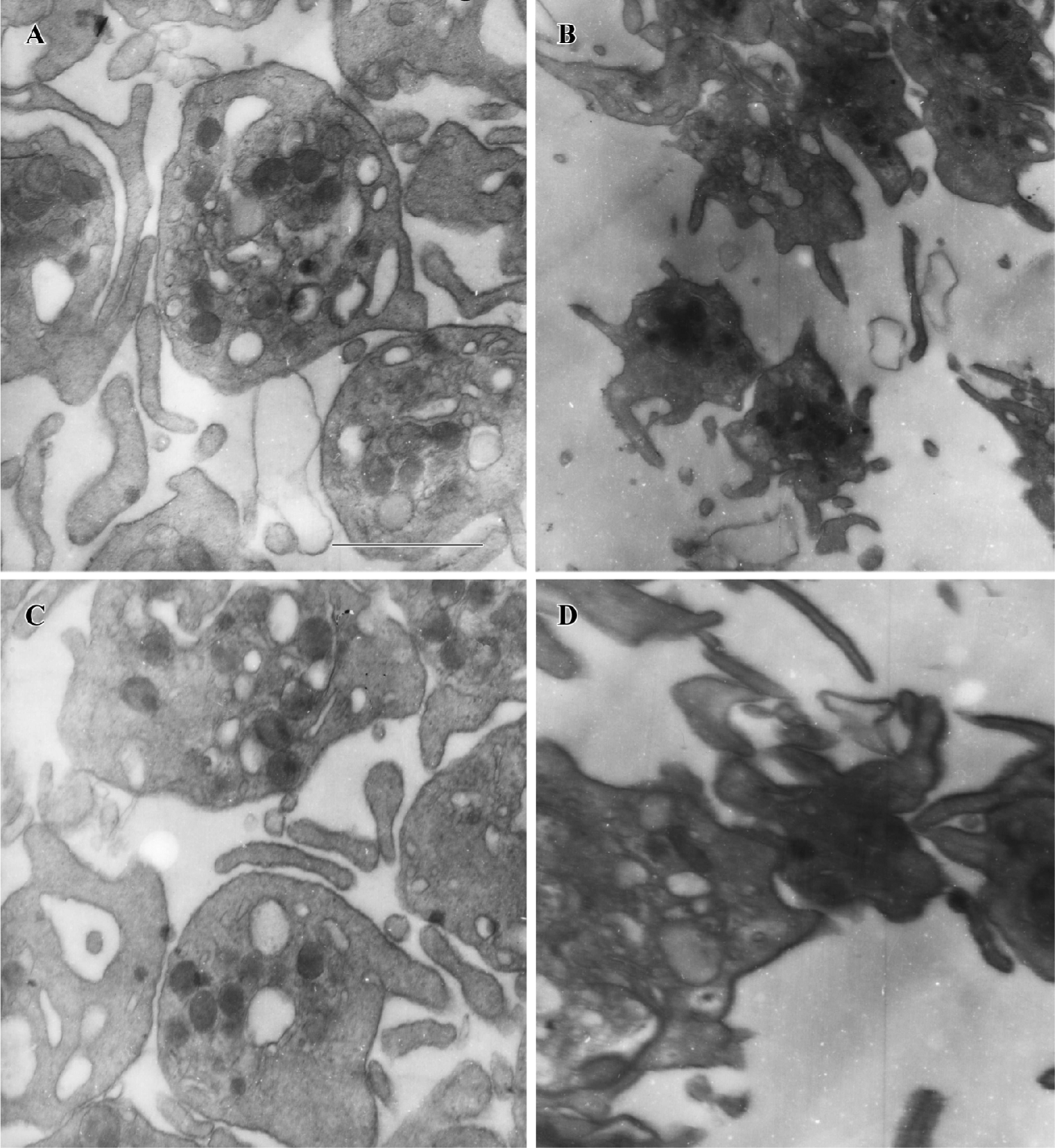
PGA1 inhibited the ultrastructural changes of platelets Stimulated platelets transform from a discoid form to a spiny spherical shape with numerous pseudopodia (shape change); simultaneously, the fibrinogen receptors that mediate aggregation are exposed. We investigated the effects of PGA1 on platelet morphology after thrombin stimulation with an electron microscope. We found that thrombin elicited robust changes in platelet morphology, including formation of numerous pseudopodia and loss of dense granules. Pretreatment with PGA1 almost completely blocked the thrombin-induced shape changes (formation of pseudopodia) and aggregation of human platelets (Figure 4).
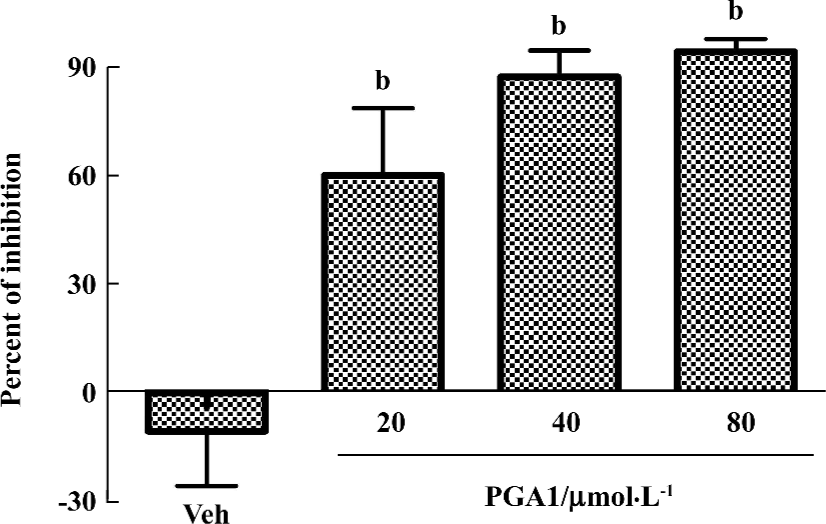
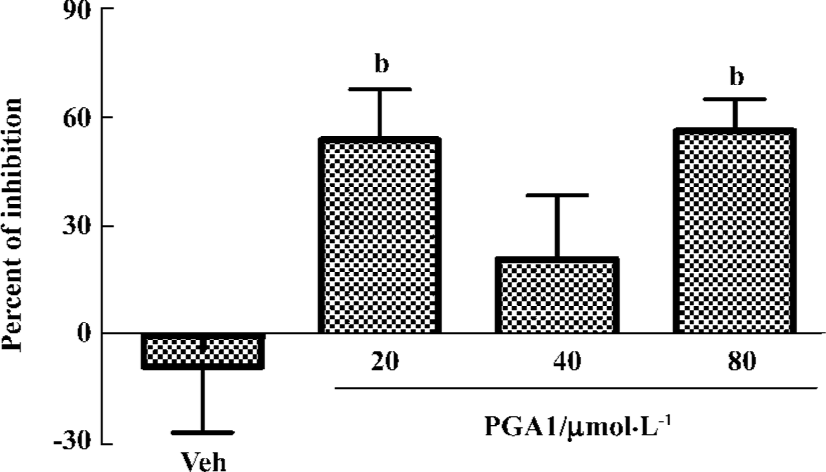
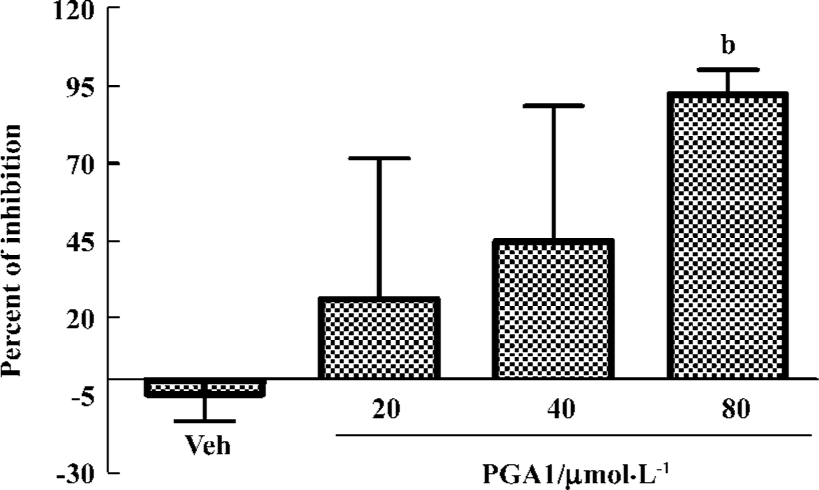
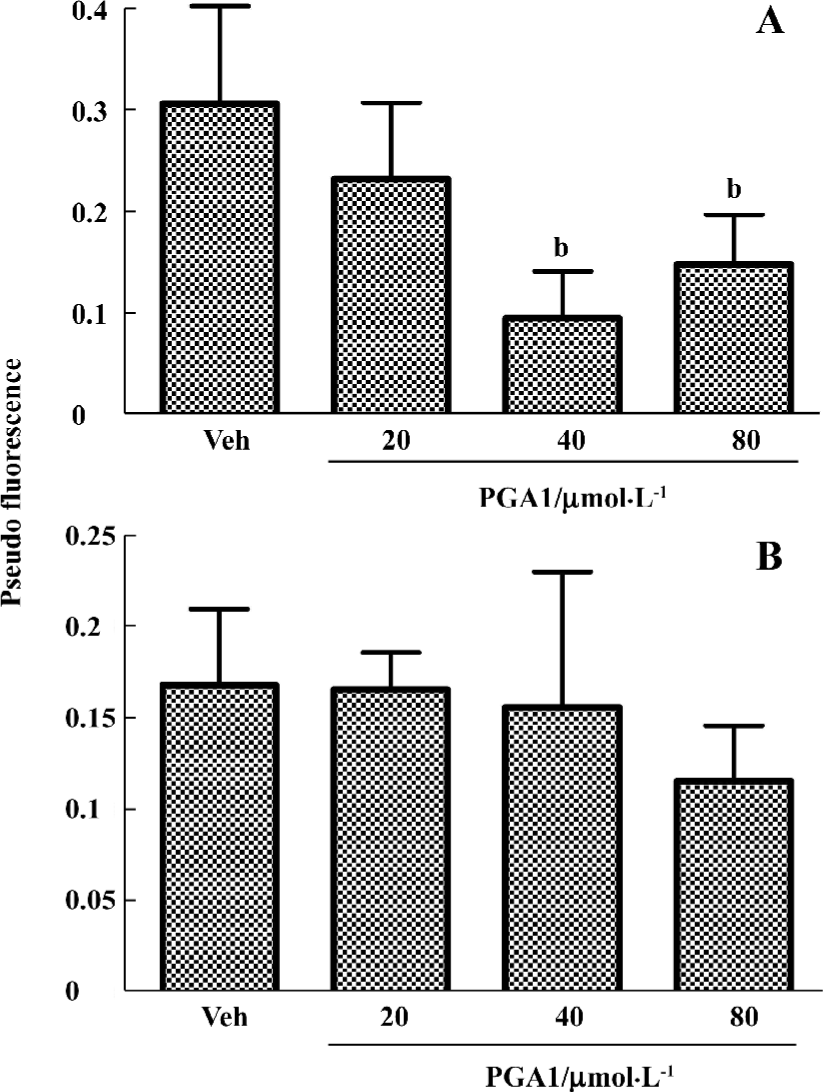
PGA1 inhibited TXA2 synthesis and calcium influx in platelets TXA2 is a potent activator of platelets. Addition of thrombin to platelets stimulated synthesis of TXB2, a stable metabolite of TXA2. PGA1 dose-dependently inhibited the production of TXB2 (P<0.05). The IC50 of PGA1 on TXB2 production was 34.1 µmol/L (P<0.05; Figure 5). Calcium is a key player in activation of platelets. PGA1 (20–80 µmol/L downregulated the increase in calcium concentration inside the platelets induced by thrombin (P<0.05; Figure 6A). When calcium was absent in the extracellular solution, we failed to observe an inhibition of calcium increase by PGA1 (Figure 6B), suggesting that PGA1 inhibited calcium influx from the extracellular space.
Discussion
Most actions of the cyclopentenone prostaglandins, including PGA2, PGA1, and PGJ2, are not mediated by binding to G-protein-coupled prostanoid receptors, but result from their direct interaction with other cellular target proteins[8]. The pharmacological actions of PGA1 with respect to its anti-inflammatory, anti-neoplastic, and anti-viral activities have been well documented. Recently, our in vivo and in vitro studies suggested that PGA1 has neuroprotective actions[12,14,15], but the mechanisms underlying its neuroprotective effects have not been fully understood.
Platelet aggregation, adhesion to endothelial cells and release of TXA2 play important roles in the formation of thrombosis. TXA2 causes constriction of blood vessels, which could further reduce blood flow in the brain. Inhibitors of platelet function are commonly used in therapeutic approaches in the treatment of brain ischemia[24,25]. Previous studies have found inconsistent results with respect to the effect of PGA1 on platelet aggregation. PGA1 has been reported to have no effect on aggregation of rabbit platelets induced by ADP, or formation of thrombus in rats[26,27], or weakly inhibited platelet aggregation at high concentrations[28]. Since platelets from different species commonly respond differently to platelet inhibitors, we thought that human platelets might be sensitive to PGA1. The present studies thus examined the effects of PGA1 on the functions of human platelets. The results showed that PGA1 significantly inhibited thrombin-induced human platelet aggregation, the release of 5-HT, and production of TXA2. Similar inhibitory results were obtained with respect to platelet aggregation induced by collagen and ADP with less potency. PGA1 inhibited the thrombin-induced increase in cytosolic [Ca2+]. These actions could be part of mechanisms by which PGA1 inhibits platelet activation. It has been well documented that in almost every step of platelet activation, calcium is required. We found that the inhibitory effect of PGA1 on rises in calcium concentration was not seen in the absence of extracellular calcium, suggesting that PGA1 may inhibit calcium influx. However, additional studies are required to confirm whether PGA1 blocks calcium entry into platelets. In our study of the effect of PGA1 on thrombin-induced 5-HT release and increases in intraplatelet calcium concentration, the medium dose of PGA1 (40 μmol/L) was less effective. The reason for this is unknown at present. Also, the effects of PGA1 on calcium entry into platelets need to be further investigated under other experimental conditions.
In addition to the above-discussed pharmacological actions, PGA1 inhibits IκB-α degradation and NF-κB activation, induces expression of heat shock proteins and activates PPAR-γ. The significance of these pharmacological actions of PGA1 with respect to the ability of PGA1 to inhibit platelets and neuronal injury in ischemia need to be further investigated.
References
- Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 2004;56:387-437.
- Satoh T, Furuta K, Tomokiyo K, Suzuki M, Watanabe Y. Designed cyclopentenone prostaglandin derivatives as neurite outgrowth-promoting compounds for CAD cells, a rat catecholaminergic neuronal cell line of the central nervous system. Neurosci Lett 2000;291:167-70.
- Piva R, Gianferretti P, Ciucci A, Taulli R, Belardo G, Santoro MG. 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis in human malignant B cells: an effect associated with inhibition of NF-kappa B activity and down-regulation of antiapoptotic proteins. Blood 2005;105:1750-8.
- Tanikawa M, Yamada K, Tominaga K, Morisaki H, Kaneko Y, Ikeda K, et al. Potent prostaglandin A1 analogs that suppress tumor cell growth through induction of p21 and reduction of cyclin E. J Biol Chem 1998;273:18522-7.
- Narumiya S, Ohno K, Fujiwara M, Fukushima M. Site and mechanism of growth inhibition by prostaglandins. II. Temperature-dependent transfer of a cyclopentenone prostaglandin to nuclei. J Pharmacol Exp Ther 1986;239:506-11.
- Piva R, Gianferretti P, Ciucci A, Taulli R, Belardo G, Santoro MG. 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis in human malignant B cells: an effect associated with inhibition of NF-kappa B activity and down-regulation of antiapoptotic proteins. Blood 2005;105:1750-8.
- Mastromarino P, Conti C, Petruzziello R, De Marco A, Pica F, Santoro MG. Inhibition of Sindbis virus replication by cyclo-pentenone prostaglandins: a cell-mediated event associated with heat-shock protein synthesis. Antiviral Res 1993;20:209-22.
- Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev 2001;21:185-210.
- Tanikawa M, Yamada K, Tominaga K, Morisaki H, Kaneko Y, Ikeda K, et al. Potent prostaglandin A1 analogs that suppress tumor cell growth through induction of p21 and reduction of cyclin E. J Biol Chem 1998;273:18522-7.
- Thomas SC, Ryan MA, Shanley TP, Wong HR. Induction of the stress response with prostaglandin A1 increases I-kappaBalpha gene expression. FASEB J 1998;12:1371-8.
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 2000;403:103-8.
- Wang X, Qin ZH, Leng Y, Wang Y, Jin X, Chase TN, et al. Prostaglandin A1 inhibits rotenone-induced apoptosis in SH-SY5Y cells. J Neurochem 2002;83:1094-102.
- Qin ZH, Wang Y, Nakai M, Chase TN. Nuclear factor-kappa B contributes to excitotoxin-induced apoptosis in rat striatum. Mol Pharmacol 1998;53:33-42.
- Zhang HL, Huang ZH, Zhu Y, Liang ZQ, Han R, Wang XX, et al. Neuroprotective effects of prostaglandin A1 in animal models of focal ischemia. Brain Res 2005;1039:203-6.
- Qin ZH, Wang Y, Chen RW, Wang X, Ren M, Chuang DM, et al. Prostaglandin A(1) protects striatal neurons against excitotoxic injury in rat striatum. J Pharmacol Exp Ther 2001;297:78-87.
- Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, et al. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem 1995;270:23975-83.
- Rossi A, Elia G, Santoro MG. Inhibition of nuclear factor kappa B by prostaglandin A1: an effect associated with heat shock transcription factor activation. Proc Natl Acad Sci USA 1997;94:746-50.
- Rossi A, Santoro MG. Induction by prostaglandin A1 of haem oxygenase in myoblastic cells: an effect independent of expression of the 70 kDa heat shock protein. Biochem J 1995;308:455-63.
- Santoro MG, Garaci E, Amici C. Prostaglandins with antiprolifera-tive activity induce the synthesis of a heat shock protein in human cells. Proc Natl Acad Sci USA 1989;86:8407-11.
- Yip HK, Liou CW, Chang HW, Lan MY, Liu JS, Chen MC. Link between platelet activity and outcomes after an ischemic stroke. Cerebrovasc Dis 2005;20:120-8.
- Tombul T, Atbas C, Anlar O. Hemostatic markers and platelet aggregation factors as predictive markers for type of stroke and neurological disability following cerebral infarction. J Clin Neurosci 2005;12:429-34.
- Luscher TF. Platelet-vessel wall interaction: role of nitric oxide, prostaglandins and endothelins. Baillieres Clin Haematol 1993;6:609-27.
- Dutta-Roy AK, Kahn NN, Sinha AK. Prostaglandin E1: the endogenous physiological regulator of platelet mediated blood coagulation. Prostaglandins Leukot Essent Fatty Acids 1989;35:189-95.
- Belayev L, Khoutorova L, Deisher TA, Belayev A, Busto R, Zhang Y, et al. Neuroprotective effect of SolCD39, a novel platelet aggregation inhibitor, on transient middle cerebral artery occlusion in rats. Stroke 2003;34:758-63.
- van Gijn J, Algra A. Aspirin and stroke prevention. Thromb Res 2003;110:349-53.
- Takano S, Itagaki S, Sakurai K, Suzuki T. Influences of prostaglandins on electrophoretic mobility and aggregation of rabbit platelets. Prostaglandins 1980;20:579-86.
- Hoshiai H, Takahashi K, Furuhashi N, Wada Y, Uehara S, Suzuki M. Cytoplasmic estrogen receptors of rat mammary glands during pregnancy and puerperium. Tohoku J Exp Med 1982;136:195-202.
- Marquis NR, Vigdahl RL, Tavormina PA. Platelet aggregation. I. Regulation by cyclic AMP and prostaglandin E1. Biochem Biophys Res Commun 1969;36:965-72.