
Increased RhoA translocation in aorta of diabetic rats1
Introduction
Diabetes mellitus is a common metabolic disease world-wide, affecting approximately 150 million people in 2000. It is predicted to affect 220 million people by 2010[1]. Diabetes and its associated complications have become a public health problem of considerable magnitude. Cardiovascular disease causes most of the excess morbidity and mortality in diabetes mellitus[1]. Although numerous therapeutic interventions available today can delay the development and progression of vascular complications associated with diabetes, there is an ongoing need for new therapeutic strategies[2]. Recent studies have demonstrated that the activation of Rho proteins appears to be a common component for the pathogenesis of hypertension and vascular proliferative disorders[3]. Functional analyses have further revealed that RhoA-dependent pathways are involved in excessive contraction, migration and proliferation associated with arterial diseases[4]. However, the role of RhoA in the development of diabetic vascular abnormalities is still unclear. The present study is designed to test the hypothesis that RhoA contributes to the development of vascular pathological changes in diabetes.
Materials and methods
Animal model Male Sprague-Dawley rats (200–250 g) were divided into 2 groups: the diabetic group (n=26) and the control group (n=26). Diabetes was induced by a single injection of streptozotocin (STZ) at a dose of 55 mg/kg (Sigma Chemical Co, St Louis, MO, USA), and the control group was injected with a 0.9% saline vehicle as described previously[5]. All injections were administered intravenously after the animals were anesthetized with sodium pentobarbital (50 mg/kg, ip). The animals were housed under identical 12-h cycling-controlled light conditions and were fed standard rat laboratory diet with free access to water. All animals were killed 3 weeks after the induction of diabetes. An additional group of rats (n=9) was used for measuring the blood pressure and heart rate for STZ-induced diabetes. The protocol for our study was approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center.
Plasma glucose, serum insulin, free fatty acid, triglycerides and cholesterol levels Blood samples were collected into heparin treated-hematocrit tubes by tail bleeding on weekly basis. Blood was separated by centrifugation and plasma glucose levels were determined using a Beckman Glucose analyzer (Beckman Instruments Inc., Fullerton, CA, USA)[6].
Three weeks after the injection of STZ, the rats were anesthetized with sodium pentobarbital (50 mg/kg, ip). Blood was drawn from the aorta just prior to removing the heart at the conclusion of the study, and the serum was separated by centrifugation. Insulin levels were determined by a radioimmunoassay kit (Linco Research Inc., St Louise, MO, USA); free fatty acid levels were measured by a diagnostic kit (Boehringer Mannheim, GmbH, Mannheim, Germany), triglycerides and cholesterol levels were measured using a diagnostic kit (Sigma Diagnostics Inc., St Louis, MO, USA)[7].
Mean arterial blood pressure and heart rate Blood pressure and heart rate were measured to assess the cardiovascular function. The experiments were conducted in 9 rats. Anesthesia was induced with sodium pentobarbital (50 mg/kg, ip), and an artery and a vein catheter were implanted under aseptic conditions as described previously[8]. After recovery from the anesthesia, the rats were placed in individual metabolic cages; the arterial catheter was filled with heparin solution (1000 USP/mL) and connected to a pressure transducer via a hydraulic swivel (Instech Laboratories, Inc, Plymouth Meeting, PA, USA). Pulsatile arterial pressure signals were amplified, sent to an analog-to-digital converter and analyzed by computer for 24 h per day using customized software. After baseline measurements, STZ (55 mg/kg, iv) was administered to all rats followed by a 3-week diabetic period.
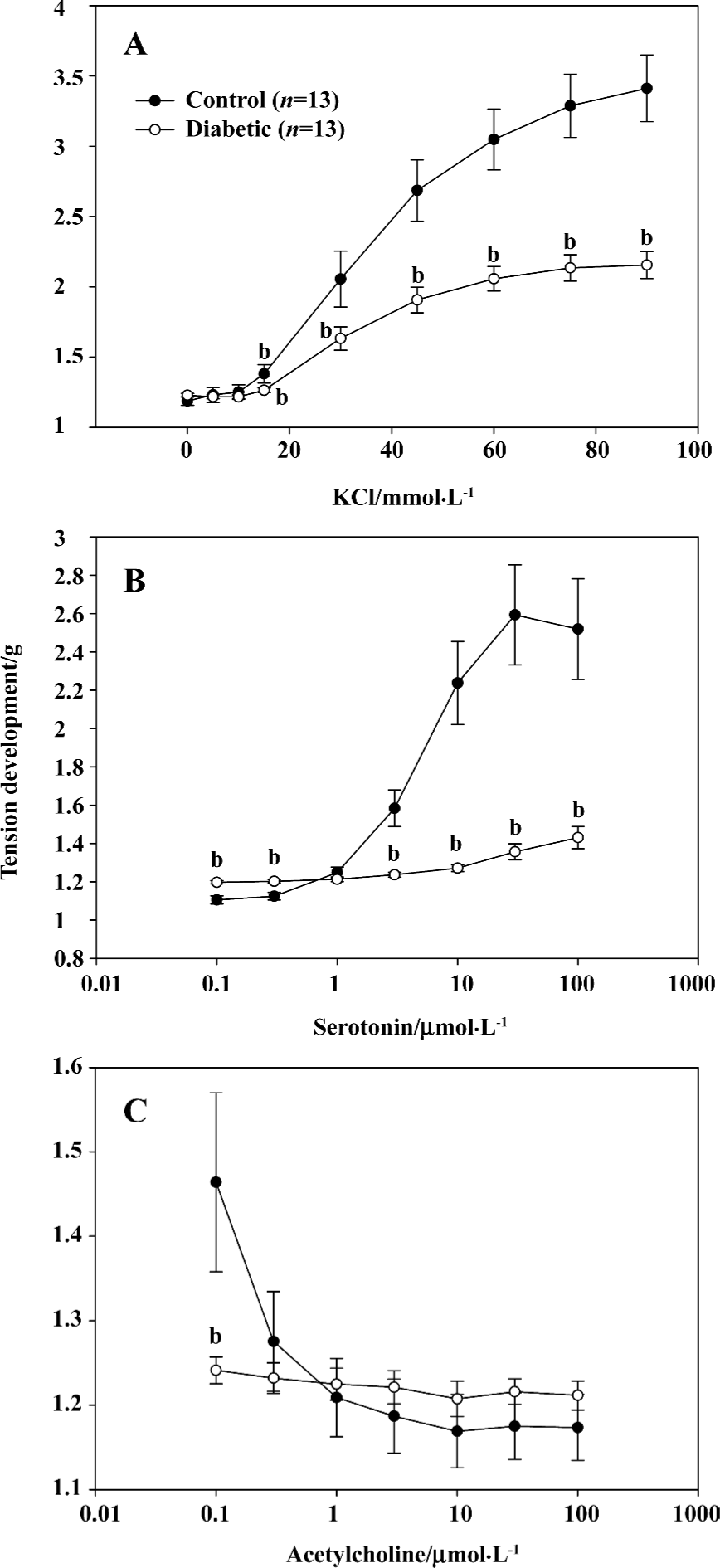
Isometric tension Contractility of the aorta from the diabetic and control rats was measured by isometric tension as previously described[9]. The aortas were removed and cut into 2-mm rings in a dissecting chamber filled with modified Krebs-Henseleit bicarbonate solution containing 120 mmol/L NaCl, 4.5 mmol/L KCl, 1 mmol/L MgSO4, 27 mmol/L NaHCO3, 1.2 mmol/L KH2PO4, 2.5 mmol/L CaCl2, and 10 mmol/L dextrose and bubbled with 95% O2–5% CO2. The rings were suspended at the resting tension of 1000 mg (Radnoti transducer, Radnoti Glass Technology, Inc, Monrovia, CA, USA) between stainless steel hooks in 10 water-jacketed tissue baths (Radnoti Glass) in modified Krebs-Henseleit biocarbonate buffer with 95% O2–5% CO2 at 37 °C. The rings were incubated for 90 min until a stable rest tension was achieved, and the solution was changed every 20 min to remove metabolites. The tissues were challenged with 60 mmol/L KCl twice at 30-min intervals before the experiment. Isometric force transducers were connected to arterial rings and contraction was recorded with an 8-channel MacLab (AdInstruments, Inc., Colorado Springs, CO, USA) and stored on a Power Macintosh computer. Concentration-response curves were established for the vasoconstriction agent serotonin, or KCl, and for the vasorelaxation agent, acetylcholine.
Protein preparation and Western blot analysis The protein expression of RhoA was examined by Western blot using total lysate as previously described[8,10]. RhoA activation was determined by its translocation from cytoplasm to membrane. In brief, the aorta tissue was isolated and washed with ice-cold PBS and then homogenized with a polytron in ice-cold buffer containing 5 mmol/L Tris-HCl, 5 mmol/L NaCl, 1 mmol/L CaCl2, 2 mmol/L egtazic acid, 1 mmol/L MgCl2, 2 dithiothreitol (DTT), and 1 µL/mL of proteinase inhibitor cocktail (Sigma, St Louise, MO, USA). Total lysate was prepared by centrifugation of homogenate at 1000×g for 15 min. To prepare cell fractions, nuclei and unlysed cells in homogenate were removed by low-speed centrifugation at 500×g for 5 min and the samples were centrifuged at 100 000×g for 60 min. The supernatant was collected as cytosolic fraction and the pellet was resuspended in the same buffer supplemented with 1% Nonidet P40 (NP-40) and then collected as membrane fraction. Protein concentrations were measured and adjusted, and Laemmli sample buffer was then added. Equal amounts of proteins were loaded in each lane, electrophoresed on 12% polyacrylamide-SDS gels and transferred to the nitrocellulose membrane. The membrane was probed with 1 μg/mL of anti-RhoA antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by horse-radish peroxidase-conjugated protein A (Zymed) at 1:10 000 dilution. Blots were exposed to film and RhoA expression in total lysate; membrane and cytosolic fractions were analyzed by densitometry.
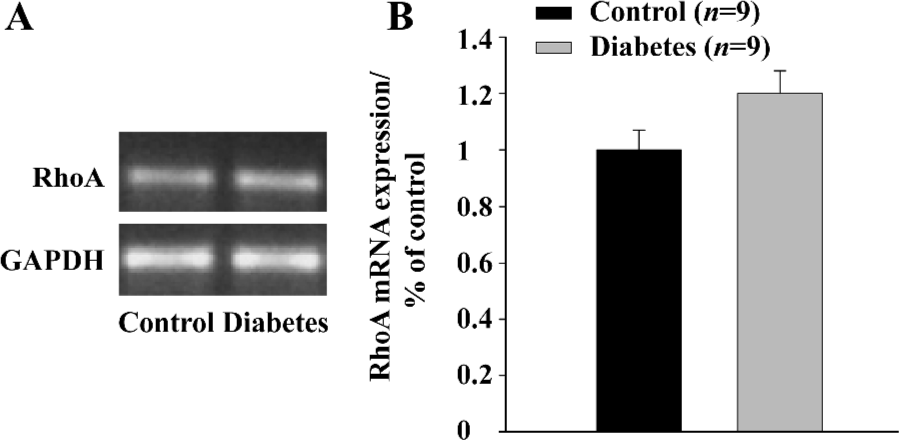
RNA isolation and reverse transcription PCR (RT-PCR) The mRNA expression of RhoA was examined by RT-PCR as described previously[11]. In brief, total RNA was isolated from the aorta with RNA STAT-60 (TEL-TEST “B”, Inc, Friendswood, TX, USA). All samples were treated with RQ1 RNase-Free DNase (Promega, Madison, WI, USA) prior to RT-PCR. cDNA was prepared from 1 μg of total RNA using SuperScript First-Strand Synthesis System for RT-PCR kit (Invitrogen, Carlsbad, CA, USA). The thermal cycle profile for PCR amplification of the 32 cycles was as follows: (1) denaturing for 30 s at 95 °C; (2) annealing primers for 30 s at 55 °C; and (3) extending the primers for 1 min at 72 °C, and using specific primers for RhoA (5' to 3' ACCAGTTCC-CAGAGGTTTATGT; 3' to 5' TTTGGTCTTTGCTGAACACT from Invitrogen, Carlsbad, CA, USA). A portion of the 10 µL of the PCR products was electrophoresed in 2% agarose gel in TBE buffer. For the quantitative analysis of the RT-PCR products, the density of the bands for mRNA was determined by a densitometer. Intensities of the bands were normalized by the intensity of the band of GAPDH and the result was expressed as the percentage of control (Figure 3).
Statistical analysis The differences between the diabetic and control groups were compared using Student t-test. Daily hemodynamic data and isometric tension were analyzed by ANOVA with repeated measures and Dunnett’s t-test. All values are expressed as mean±SEM. The level of significance was set at P<0.05.
Results
Diabetes was confirmed by hyperglycemia after STZ-injection (plasma glucose was 624.0±15.8 mg/dL in diabetes vs 120.0±3.7 mg/dL in control, P<0.01, Student t-test). Diabetic animals had lower body weights (241.1±13.4 g) than those in the control group (352.3±24.7 g, P<0.01). Serum insulin levels were markedly lower in the diabetic animals (5.10±0.69 µU/mL) compared with the control group (32.80±6.65 µU/mL, P<0.01), and lipid levels increased in the diabetic rats [Free amino acid (FAA) 0.59±0.08 mmol/L, TG (4.48±0.90) mg/mL, cholesterol (0.89±0.06) mg/mL] when compared with the rats in the control group [FAA 0.21±0.03 mmol/L, TG (0.658±0.081) mg/mL, cholesterol (0.675±0.042) mg/mL, P<0.01, res-pectively].
Hemodynamics Heart rate averaged 408.0±2.0 beats/min during the baseline period and decreased markedly (P<0.05, Student t-test) after the onset of diabetes (350±15 beats/min), which is consistent with previous reports[12]. The mean arterial pressure averaged 95.0±0.2 mmHg during baseline, and the average of 97.0±0.4 mmHg during the diabetic period was not significantly different (P>0.05, Student t-test).
Contraction and relaxation of STZ-induced diabetic aorta KCl produced a concentration-dependent contraction in the aortas of both the diabetic and control rats. However, the KCl-induced contraction of the aorta in the diabetic rats was significantly reduced compared with that in the control rats (Figure 1A), which is consistent with previous reports[13–15]. Serotonin-induced contraction of the aorta in diabetic rats was almost abolished compared with the control rats (Figure 1B), which supports the findings of Reyes-Toso[16] and Hattori[17]. Diabetes prohibited acetylcholine-induced relaxation in rat aortas (Figure 1C) which is consistent with other published reports[13].
RhoA protein expression and activation in the aorta (Western blot) RhoA protein expression in the total lysate extracted from the aorta tissue was not significantly different (P>0.05) between the diabetic and control groups (Figure 2A), and was consistent with the finding of RhoA mRNA expression. These data indicate that total RhoA expression did not change in the aorta of STZ-induced diabetic animals.
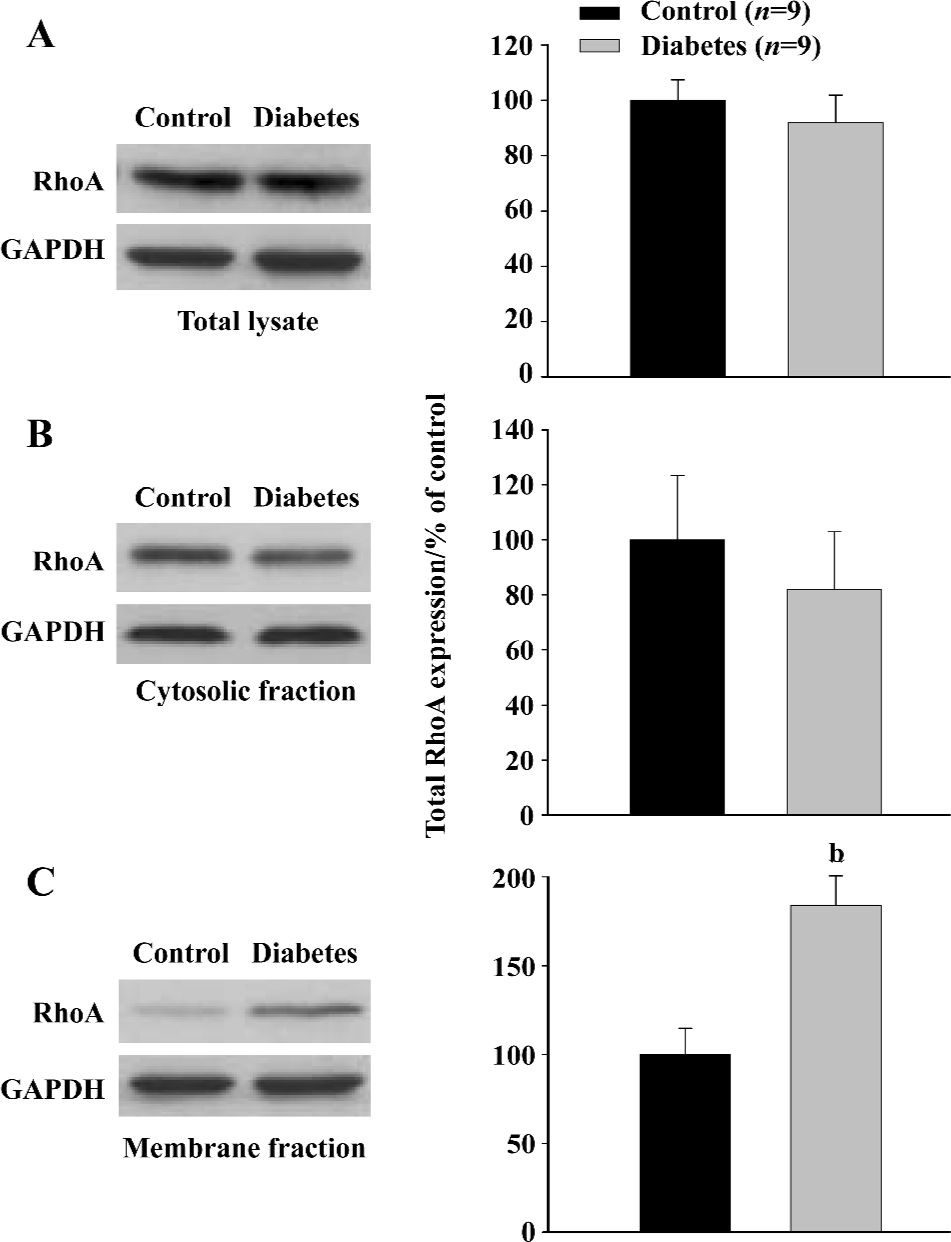
Increases in translocation from the cytosolic to the membrane fraction are associated with RhoA activation[3]. To investigate the effect of STZ-induced diabetes on the activation of RhoA, we measured membrane-associated RhoA and cytosolic RhoA in the aortic tissue in diabetic and control rats. Even though no significant change was found in the cytosolic RhoA expression (Figure 2B), there was a 90% increase (P<0.05, Student t-test) of membrane-associated RhoA expression in the diabetic group compared with the control group (Figure 2C). The ratio (calculated from the intensity) of RhoA protein in membrane/RhoA protein in cytoplasm is significantly higher in diabetic rats (0.73±0.08) than that in control rats (0.39±0.07, P<0.05), indicating an increased RhoA translocation in aorta of diabetic rats.
These data indicate that RhoA activation was markedly upregulated in the aorta of STZ-induced diabetic rats.
RhoA mRNA expression in aorta (RT-PCR) To determine whether RhoA transcription in the aorta was affected by STZ-injection, RhoA mRNA expression was measured by quantitative RT-PCR. Figure 3 shows that there was no significant deference in RhoA mRNA expression in the aortic tissue of diabetic rats compared with the control rats (P>0.05).
Discussion
This study has demonstrated the following observations: (1) there is significantly increased RhoA activation in the aorta of diabetic rats; (2) concentration-dependent contraction induced by KCl or serotonin was markedly reduced in the aorta of diabetic rats as well as the relaxation induced by acetylcholine; and (3) there is no significant difference in the mean arterial blood pressure between the control and diabetic rats suggesting that the change in RhoA activation occurred before the onset of severe functional abnormalities. Our data suggest that RhoA might be involved in the etiology of diabetic vascular dysfunction.
Diabetes mellitus is a major and increasing health problem worldwide. One of the most serious consequences of diabetes is the development of diabetic angiopathy, which includes cardiovascular disease, neuropathy, retinopathy and nephropathy[2]. Adults with diabetes are at a 2−4-fold increased risk of cardiovascular disease relative to those without diabetes. Cardiovascular disease accounts for up to 80% of premature excess mortality in diabetic patients[1]. Studies on animals suggest that diabetes-induced vascular complications may result from abnormalities of the contractile and regulatory proteins in diabetes[13,15], however, the pathogenesis of diabetic vascular dysfunction remains unclear.
Our data show that the maximum contraction of the aorta induced by KCl and serotonin in diabetic rats is markedly reduced compared with age-matched control rats, indicating the contraction induced by release of Ca2+ from intracellular stores in response to serotonin as well as the contraction induced by Ca2+ influx in response to KCl are diminished in aortic tissues of diabetic rats. We also found that diabetes abolished acetylcholine-induced relaxation. All these observations indicate a dysfunctional smooth muscle contractile mechanism in STZ-induced diabetic rats. These observations are consistent with the previous reports that diabetes reduces contraction in response to KCl, CaCl2, endo-thelin-1, serotonin, and decreases relaxation in response to acetylcholine in aortas of diabetic animals[13–15,17].
RhoA is a GTPase that has been shown to play a key role in both actin stress fiber formation and focal adhesion complex assembly in fibroblasts[19]. RhoA belongs to the Rho family, which includes Rho, Rac, and Cdc42. There are 3 isoforms of Rho (A, B, and C). Rho shuttles between the active GTP-bound form on the cell membrane and the inactive GDP-bound form in the cytoplasm. Direct targets for Rho have been sought by screening for molecules selectively binding to, or activated by GTP-Rho[20,21]. Among the 3 isoforms, RhoA is the most ubiquitously and abundantly expressed in the body, and has been extensively studied. Recent studies demonstrate that RhoA is an important target in the cell signaling pathway and that RhoA plays a pivotal role in cell function. In smooth muscle cells, Hirata et al[22] first reported that RhoA was involved in the myofilament Ca2+-sensitization. RhoA activates Rho kinase, which in turn phosphorylates and inactivates myosin light chain (MLC)-phosphatase and subsequently enhances MLC-phos-phorylation, resulting in vasoconstriction[23,24]. The activated Rho-mediated pathway has been linked to hypertension[25], coronary and cerebral vasospasm [26,27], and heart failure in several animal models[28].
Recent studies suggest that RhoA/Rho kinase play an important role in the regulation of cavernosal smooth muscle tone and that changes in this pathway could contribute to erectile dysfunction in diabetic patients and STZ-induced diabetic rats[18,29–30]. In GK-diabetic rats, diabetes causes elevation in Rho kinase activity in vascular smooth muscle cells leading to myosin-bound subunit phosphorylation, which further inactivates myosin-bound phosphates causing excessive contractility of vascular smooth muscle cells. Insulin stimulates myosin-bound phosphates in part by reducing the phosphorylation of its regulatory subunit, myosin-bound subunit via Rho kinase inhibition[31]. In obese Zucker rats, endothelium-dependent vasorelaxation was impaired and Ca2+ sensitization of contraction was augmented in the blood vessels. The enhanced vasoconstriction was abolished when the aorta of obese Zucker rats was pre-incubated with the Rho kinase inhibitor Y-27632[32].
The overall observations of our experiments from STZ-induced diabetic rats are consistent with these previous findings. We found that impaired vascular sensitivity was associated with enhanced RhoA activity indicated by increased RhoA translocation in the aortic tissues. High levels of RhoA at membrane portion may potentiate contractility and render diabetic aorta remaining in a partially contractile state as has been observed by others[32]. This increased basal contractile level in the diabetic aorta led to a reduction of the maximum contraction when compared with the basal tension. Indeed, AngII (1×10-5 mol/L) produced approximately 0.1 g tension in the aorta of the lean Zucker rats (control), but produced only about 0.02 g tension in the obese Zucker rats[32]. If the maximum tension is plotted against the baseline tension, then the contractions of aortas in response to KCl and serotonin were markedly reduced in the STZ-induced diabetic rats as observed in the present study. The relaxation in response to acetylcholine was abolished by diabetes indicating either an impaired endothelium[13–15,17] or a higher contractile stage of smooth muscle cells due to the activation of RhoA and Rho-kinase[32]. RhoA mRNA expression and total RhoA protein expression in the aorta did not increase in the diabetic rats, and there was no significant difference in arterial blood pressure between diabetic and control rats, suggesting that the activation of RhoA alone in the early stage of diabetes may have been prior to the severe functional changes of the arterial vessels and possible structural injury.
Our observation supports previously published findings that RhoA activation is enhanced in diabetic aortic tissues which can contribute to the vascular dysfunction in diabetic rats. Future studies should be performed to demonstrate that the increase of RhoA activity correlates with the impaired vascular contractile, and relaxation responses and the inhibitors of RhoA/Rho kinase should be employed.
References
- Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci (Lond) 2005;109:143-59.
- Ostergaard J, Hansen TK, Thiel S, Flyvbjerg A. Complement activation and diabetic vascular complications. Clin Chim Acta 2005;361:10-9.
- Ren J, Fang CX. Small guanine nucleotide-binding protein Rho and myocardial function. Acta Pharmacol Sin 2005;26:279-85.
- Pacaud P, Sauzeau V, Loirand G. Rho proteins and vascular diseases. Arch Mal Coeur Vaiss 2005;98:249-54.
- Ryan EA, Tobin BW, Tang J, Finegood DT. A new model for the study of mild diabetes during pregnancy. Syngeneic islet-transplanted STZ-induced diabetic rats. Diabetes 1993;42:316-23.
- Tang J, Neidigh JL, Cooksey RC, McClain DA. Transgenic mice with increased hexosamine flux specifically targeted to beta-cells exhibit hyperinsulinemia and peripheral insulin resistance. Diabetes 2000;49:1492-9.
- Massey AR, Miao L, Smith BN, Liu J, Kusaka I, Zhang JH, et al. Increased RhoA translocation in renal cortex of diabetic rats. Life Sci 2003;72:2943-52.
- Tang J, Fitzgerald SM, Boughtman BN, Cole SW, Brands MW, Zhang JH. Decreased RhoA expression in myocardium of diabetic rats. Can J Physiol Pharmacol 2005;83:775-83.
- Miao L, Dai Y, Zhang J. Mechanism of RhoA/Rho kinase activation in endothelin-1-induced contraction in rabbit basilar artery. Am J Physiol Heart Circ Physiol 2002;283:H983-9.
- Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, et al. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab 2004;24:1133-45.
- Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil GD, et al. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem 2005;94:1342-50.
- Tomlinson KC, Gardiner SM, Hebden RA, Bennett T. Functional consequences of streptozotocin-induced diabetes mellitus, with particular reference to the cardiovascular system. Pharmacol Rev 1992;44:103-50.
- Mahmoudian M, Behnaz F, Rezaei E. Diabetes-induced changes in the contractility of the aorta and pA2 of nifedipine in the rat. Acta Diabetol 1996;33:114-7.
- Fulton DJ, Hodgson WC, Sikorski BW, King RG. Attenuated responses to endothelin-1, KCl and CaCl2, but not noradrenaline, of aortae from rats with streptozotocin-induced diabetes mellitus. Br J Pharmacol 1991;104:928-32.
- Kawasaki H. Pharmacological studies on alterations in contractile reactivity in aortas isolated from experimental diabetic rats. Hokkaido Igaku Zasshi 1997;72:649-65.
- Reyes-Toso CF, Roson MI, Albornoz LE, Damiano PF, Linares LM, Cardinali DP. Vascular reactivity in diabetic rats: effect of melatonin. J Pineal Res 2002;33:81-6.
- Hattori Y, Kawasaki H, Kanno M, Fukao M. Enhanced 5-HT2 receptor mediated contractions in diabetic rat aorta: participation of Ca2+ channels associated with protein kinase C activity. J Vasc Res 1995;32:220-9.
- Bivalacqua TJ, Champion HC, Usta MF, Cellek S, Chitaley K, Webb RC, et al. RhoA/Rho-kinase suppresses endothelial nitric oxide synthase in the penis: a mechanism for diabetes-associated erectile dysfunction. Proc Natl Acad Sci USA 2004;101:9121-6.
- Mackay DJ, Hall A. Rho GTPases. J Biol Chem 1998;273:20685-8.
- Narumiya S. The small GTPase Rho: cellular functions and signal transduction. J Biochem (Tokyo) 1996;120:215-28.
- Niiro N, Nishimura J, Sakihara C, Nakano H, Kanaide H. Up-regulation of Rho A and Rho-kinase mRNAs in the rat myometrium during pregnancy. Biochem Biophys Res Commun 1997;230:356-9.
- Hirata K, Kikuchi A, Sasaki T, Kuroda S, Kaibuchi K, Matsuura Y, et al. Involvement of Rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. J Biol Chem 1992;267:8719-22.
- Noda M, Yasuda-Fukazawa C, Moriishi K, Kato T, Okuda T, Kurokawa K, et al. Involvement of Rho in GTP gamma S-induced enhancement of phosphorylation of 20 kDa myosin light chain in vascular smooth muscle cells: inhibition of phosphatase activity. FEBS Lett 1995;367:246-50.
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996;273:245-8.
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997;389:990-4.
- Shimokawa H, Seto M, Katsumata N, Amano M, Kozai T, Yamawaki T, et al. Rho-kinase-mediated pathway induces enhanced myosin light chain phosphorylations in a swine model of coronary artery spasm. Cardiovasc Res 1999;43:1029-39.
- Miyagi Y, Carpenter RC, Meguro T, Parent AD, Zhang JH. Upregulation of Rho A and Rho kinase messenger RNAs in the basilar artery of a rat model of subarachnoid hemorrhage. J Neurosurg 2000;93:471-6.
- Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, Ross J Jr, et al. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest 1999;103:1627-34.
- Andersson KE. Erectile physiological and pathophysiological pathways involved in erectile dysfunction. J Urol 2003;170:S6-13.
- Chang S, Hypolite JA, Changolkar A, Wein AJ, Chacko S, Disanto ME. Increased contractility of diabetic rabbit corpora smooth muscle in response to endothelin is mediated via Rho-kinase beta. Int J Impot Res 2003;15:53-62.
- Sandu OA, Ragolia L, Begum N. Diabetes in the Goto-Kakizaki rat is accompanied by impaired insulin-mediated myosin-bound phosphatase activation and vascular smooth muscle cell relaxation. Diabetes 2000;49:2178-89.
- Nishimatsu H, Suzuki E, Satonaka H, Takeda R, Omata M, Fujita T, et al. Endothelial dysfunction and hypercontractility of vascular myocytes are ameliorated by fluvastatin in obese Zucker rats. Am J Physiol Heart Circ Physiol 2005;288:H1770-6.