
Effect of 7-hydroxystaurosporine on glioblastoma cell invasion and migration1
Introduction
Protein kinase C (PKC) is a family of serine/threonine protein kinases that transduce signals for tumorigenesis, tumor cell invasion, and metastasis, and has thus been newly targeted for use in cancer treatment[1]. Activation of PKC augments tumor cell metastatic potential, whereas suppression of PKC activity through PKC inhibitors reduces tumor cell invasion and migration[1,2]. 7-Hydroxystaurosporine (UCN 01) is a selective PKC inhibitor derived from the non-selective protein kinase inhibitor staurosporine[3]. A number of studies, including some conducted in our own labora-tories, have revealed that UCN-01 holds promise for use as a single agent or in combination with other chemotherapeutic agents, such as camptothecin, 5-fluorouracil, tamoxifen, and ionizing radiation, in inhibiting tumor cell growth in vitro and in vivo[3–9].
Our previous studies corroborate other studies, which indicate that the anti-tumor activities of UCN-01 are associated with arrest of cell cycle progression, including G1/S and/or G2/M[4,5], apoptosis induction[5–7,10], and the inhibition of DNA repair[11,12]. Most notably, UCN-01 abrogates the S or G2 arrest caused by chemotherapeutic agents[4]. It has also been found that UCN-01 inhibits microvessel formation (angiogenesis), which is required for tumor formation and growth[13,14]. Taken together, these studies indicate that UCN-01 is a profound anti-tumor agent in several different tumors, including glioma. Moreover, UCN-01 has entered Phase I trials as a single agent in the USA and Japan[3,15].
In the present study, the effects of UCN-01 on cell growth, invasion, and migration were investigated in U-87MG, an invasive human glioblastoma cell line. The purpose of this study is to explore whether UCN-01 can be used as a new therapeutic agent in the treatment of glioblastoma growth and invasion.
Materials and methods
Cell culture and chemicals The human glioblastoma cell line U-87MG was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained as monolayer cultures in D-MEM supplemented with 10% fetal calf serum (FCS), glutamine 2 mmol/L, streptomycin 100 mg/L, and 100 kU/L benzylpenicillin G (BioWhittaker, Walkersville, MD, USA). Cell number and viability were determined by staining a small volume of cell suspension with 0.4% trypan blue saline solution and examining the cells using a haemocytometer. The doubling time of U-87MG cell was approximately 30 h under our culture conditions. UCN-0 was obtained from the Laboratory of Molecular Pharmaco-logy, Division of Basic Science, NCI, NIH (Bethesda, MD, USA) and stored at -20 oC as a 10 mmol/L stock solution in 20% Me2SO. Phorbel 12-myristate-13-acetate (PMA) and ethanol (EtOH) were purchased from Sigma Chemical Company (St Louis, MO, USA). PMA was dissolved in 20% Me2SO and frozen at -20 oC for storage purposes. UCN-01, PMA, and EtOH were further diluted in 2% medium prior to use.
Expression vectors and transfection A full-length BRCA1 or PTEN cDNA was expressed in a pCMV-Tag2B vector (Stratagene, La Jolla, CA, USA), which allows for the expression of proteins with an N-terminal FLAG sequ-ence, as described previously[16]. Transfection of BRCA1 or PTEN was performed using a transfection reagent, Lipofect-Amin 2000, according to the manufacturer’s instructions (Invitrogene, Gaithersburg, MD, USA) as described previously[16]. The efficiency of gene transfer was determined by a β-galactosidase assay using co-transfection with plasmid pRSV-β-gal.
Measurement of PKC activity Aliquots of 4×106 untreated and treated cells were washed with ice-cold Dulbecco’s phosphate-buffered saline (PBS), suspended in a homogenization buffer (0.05 mol/L Tris-HCl, pH 7.5, containing EDTA 5 mmol/L, EGTA 10 mmol/L, β-mercaptoethanol 0.6 g/L, leupeptin 10 mmol/L, PMSF 1 mmol/L), and broken by probe sonication. The nuclei and unbroken cells were removed using low-speed spin, and the resulting supernatant was spun at 100 000×g for 30 min. The nuclear pellet was resuspended in HB and was referred to as a whole cell lysate. The 100 000×g supernatant was termed the soluble or cytosolic fraction. The 100 000×g pellet was washed with homogenization buffer, suspended by probe sonication in homogenization buffer containing 0.1% Triton X-100, incubated on ice for 1 h, and spun again at 100 000×g. The resulting supernatant was termed the particulate or membrane fraction.
Total PKC activity in the three fractions was estimated using a commercially available assay system (Amersham Life Science, Arlington Heights, IL, USA) according to the manufacturer’s instructions. This assay is based on the PKC-catalyzed transfer of the 32P-phosphate group from [γ-32P]ATP into a PKC-specific peptide substrate (amino acids 65-658 of the EGF receptor with the phosphorylation site on Thr-654) in the presence of Ca2+ phosphatidylserine, and phorbol 12-myristate 13-acetate. All 3 fractions were diluted with 0.05 mol/L Tris-HCl (pH 7.5), and 25 μg of each were added to equal volumes of reaction solution (consisting of Tris-HCl 0.05 mol/L, CaCl2 1.5 mmol/L, dithiothreitol 7.5 mmol/L, PKC peptide substrate 45 mmol/L, and dioleoyl-glycerol 82 mmol/L) as provided by the manufacturer. Reactions were carried out at 37 oC for 15 min. After 15 min, the reactions were stopped using a stop reagent (provided by the manufacturer); one hundred microlitre of each were blotted on glass fiber filters and extensively washed with 0.1% orthophosphoric acid. The bound radioactivity was counted using liquid scintillation spectroscopy. The protein content in each of the 3 fractions was measured. Radioactivity values resulting from the phosphorylation of endogenous substrates were subtracted from all determinations. Reactions were performed in triplicate and the values were averaged. Each assay was performed at least 3 times.
Measurement of cell viability Cell viability, an indicator of cytotoxicity, was evaluated using an MTT assay as described previously[17]. Four sets of experiments were performed in 10 wells for each treatment.
In vitro invasion assay In vitro invasion assay was performed with a modified Boyden chamber[17]. The surfaces of filter (0.8 μm pore size) were coated with 25 μg Matrigel of uniform thickness for 1 h at room temperature. The uniformity of the coating was checked by Coomassie blue staining and low-power microscope observation. The lower chamber was filled with 10% FCS medium containing fibronectin (16 μg per chamber) as the chemoattractant. Cells (1×108 cells/L) re-suspended in the medium containing 2% FCS and 50 or 100 nmol/L UCN-01 were carefully transferred onto the upper surface of the filters in the chamber. After a 48-h incubation, the filter was gently removed from the chamber. The cells on the upper surface were removed with a cotton swab, cells that had passed through the Matrigel and attached themselves to the lower surface of the filter were fixed, stained with hematoxylin and eosin, and counted in 15 randomly selected microscopic fields (×400) per filter. Experiments were performed at least 3 times, independently. In the trials for determining the effect of UCN-01 on the PMA- and EtOH-promoted cell invasion, fibronectin was not added to the lower chamber.
Scratch wound assay The spreading and migration capabilities of U-87MG cells were assessed using a scratch wound assay[17] which measures the expansion of a cell population on surfaces. The cells were seeded into 6-well tissue culture dishes at a concentration of 2.5×105 cells and cultured in medium containing 10% FCS to nearly confluent cell monolayers, which were then carefully wound using 1-mL sterile pipette tips. Any cellular debris was removed by washing with PBS. The wounded monolayers were then incubated in 10% FCS medium containing UCN-01 (50 or 100 nmol/L) for 24 h or 48 h, then photographed under a light microscope (×200). The experiments were repeated in quadruplicate wells at least 3 times.
Immunoblot assay Protein expression was assessed using an immunoblot assay as described previously[16]. A monoclonal E-cadherin antibody was purchased from Transduction Laboratories (Lexington, KY, USA). A mouse monoclonal anti-FLAG antibody M2 (Stratagene, La Jolla, CA, USA) was used to detect expression of the FLAG-tagged BRCA1 and PTEN proteins. Equal protein loading and the protein transfer were confirmed by immunoblotting for the determination of α-actin protein using a polyclonal α-actin antibody (I-19, Santa Cruz, Hercule, CA, USA) on the same Western blots stripped. A colored marker (Bio-Rad Labora-tories, Hercules, CA, USA) was used as a molecular size standard.
Reverse transcription-polymerase chain reaction (RT-PCR) mRNA was assayed by RT-PCR as described in previous studies[16]. cDNA was synthesized from 2 µg of total RNA in a 30-µL reaction mixture containing 5×reverse transcriptase reaction buffer (Life Technologies, Inc, Gaithers-burg, MD, USA), dNTP 200 µmol/L , 100 µmol/L solution of primers, 50 units of RNasin (Promega, Madison, WI, USA), dithiotheithol 10 mmol/L, and 100 units of reverse transcriptase (Life Technologies, Inc). The mixture was incubated at 37 °C for 60 min, heated to 95 °C for 10 min, and then chilled on ice. PCR was carried out in a 50-µL volume containing 10–20 ng cDNA, chelating buffer (Perkin-Elmer/Cetus, Norwalk, CT, USA), 20 µmol/L dNTP mixture, 1.5 units of Taq DNA polymerase (Perkin-Elmer/Cetus), and 0.5 µmol/L of the following E-cadherin-specific primer pairs: 5'-CAATCTCA-AGCTCATGG-3' (forward) and 5'-CCATTCGTTCAAGTA-GTC-3' (backward). The PCR was processed at 94 °C for 1 min, 54 °C for 1 min, and 72 °C for 1 min. To ensure that the RNA was of sufficient purity to undergo RT-PCR, a PCR assay using primers specific for the β-actin gene cDNA was performed for each sample through the same PCR process. β-Actin specific primer pairs were as follows: 5'-GTC AAC GGA TTT GGT CTG TAT T-3' (forward); 5'-AGT CTT CTG GGT GGC AGT GAT-3' (backward). Semi-quantitative PCR conditions for E-cadherin and β-actin are 28 cycles and 22 cycles, respectively. The PCR products were electrophoresed on a 5% nondenaturing polyacrylamide gel. The gel was then dried and exposed to an imaging plate, and the radioactivity was determined using a Bioimage Analyzer (Bas1000; Fuji, Kanagawa, Japan).
Cell-cell aggregation assay Cells were harvested using HBSS buffer containing 0.01% trypsin and centrifuged at 100 000×g for 5 min. Cell pellets were re-suspended in a HCMF buffer (NaCl 160 mmol/L, Na2HP4 0.6 mmol/L, 0.1% w/v glucose, and 0.01% HEPES, pH 7.4) containing CaCl2 4 mmol/L at a cell density of 1×105 cell/ml. Total 1 mL aliquots of a single cell suspension were transferred to microcentrifuge tubes. UCN-01 (50 or 100 nmol/L), anti-E-cadherin antibody (0.5 mg/L, Transduction Laboratories, Lexington, KY, USA), or a combination of both was added. The cell cultures were then put on a shaker maintained at 37 oC for 60 min and 0.02 mL aliquots from all cell cultures were removed and fixed in 0.060 mL of glutaraldehyde. The total number of particles (that is, cells or aggregates) were counted by two investi-gators, using a haemocytometer. The degree of aggregation is represented by the aggregation index calculated by Nt=60/Nt=0, where Nt=0 is the number of single cells before the incubation and Nt=60 is the number of single cells after the incubation for 60 min. Nt=x60/Nt=0>1=no cell-cell adhesion; Nt=60/Nt=0<1=specific cell-cell adhesion.
Statistical analysis Each assay was performed at least 3 times. Statistical significance for the results was assessed using Student’s t-test. P<0.05 suggests a statistically significant difference.
Results
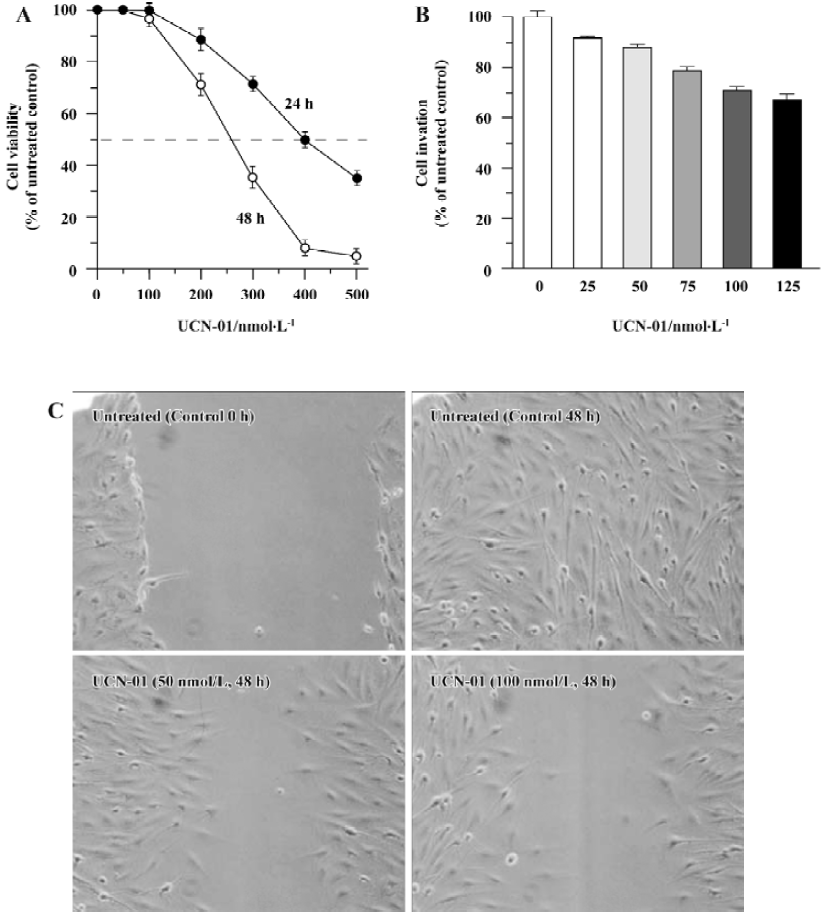
Effect of UCN-01 on proliferation of U-87MG cells Using an MTT assay, we determined the cell viability of U-87MG glioblastoma cells after treatment with various doses of UCN-01 for 24 or 48 h. As shown in Figure 1A, exposure to UCN-01 resulted in a loss of cell viability in a concentration- and time-dependent manner. ID50, a dose at which 50% of the cells lost their viability, was approximately 400 nmol/L and 260 nmol/L after the 24-h and 48-h treatments, respectively (P<0.01). At UCN-01<100 nmol/L doses, no significant effect on cell viability was observed at any exposure time points (P>0.01). UCN-01 doses of <100 nmol/L were thus selected for use in further studies. We also determine the cytotoxicity of UCN-01.
Effect of UCN-01 on PKC activity of U-87MG cells Subconfluent cells were left untreated or treated with 100 nmol/L UCN-01 for 4 h and then harvested for assay of PKC activity . The membranous fractions (404±23 pmol 32P/min, P<0.01) and cytosolic factions (114±46 pmol 32P/min, P<0.01) were significantly inhibited in the cells treated with UCN-01 compared to the untreated control cells (885±42 pmol 32P/min for the membranous fractions, 289±65 pmol 32P/min for the cytosolic fractions). Similarly, reduced PKC activity was also observed after treatment with 50 nmol/L UCN-01 (data not shown).
Effect of UCN-01 on invasion and motility of U-87MG cells Cell invasion and migration are crucial processes in tumor metastasis. A modified Boyden chamber assay was performed in order to determine the ability of U-87MG cells to invade through biological matrices in vitro, based on the percentage of cells able to penetrate the reconstituted basement membrane-coated filters and attach to the lower surface of filter. As shown in Figure 1B, the invasion capacity of U-87MG cells was markedly inhibited when UCN-01 was present in the upper chamber; a reduction of approximately 12% and 25% (P<0.01) was observed for 50 and 100 nmol/L UCN-01 after 48-h treatment, respectively. A similar inhibition of cell invasion activity by UCN-01 was also observed in U-373, another glioblastoma cell line (data not shown).
To examine whether UCN-01 anti-invasion potential is associated with its ability to suppress cell spreading and migration, the effect of UCN-01 on U-87MG cell motility was also analyzed using a scratch wound assay. As demonstrated by the representative fields shown in Figure 1C, 50 nmol/L UCN-01 markedly inhibited the flattening and spread of both cell lines along the edges of the wound compared to the untreated control cells. When this experiment was repeated using a UCN-01 dosage of 100 nmol/L instead of 50 nmol/L, the anti-migration observed was more significant.
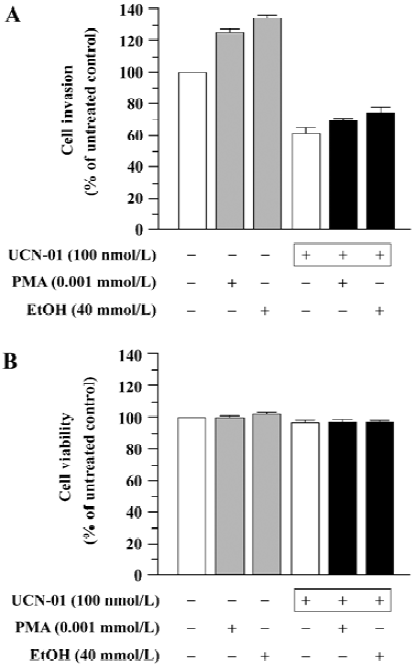
Effect of UCN-01 on PMA- and EtOH-stimulated cell invasion We determined the effect of UCN-01 on the stimulation of cell invasion caused by PMA and EtOH, two PKC activators. U-87MG cells spontaneously penetrated through an artificial basement membrane, and the number of invading cells significantly increased when PMA or EtOH was included in the cell suspension [invasion index 58±4 (PMA), 62±6 (EtOH) versus the untreated control, 46±3; P<0.01]. However, both spontaneous invasion and PMA- or EtOH-promoted cell invasion were markedly inhibited by UCN-01(Figure 2A). The ability of UCN-01 to suppress PMA- and EtOH-promoted cell invasion could not be attributed to increased cytotoxicity, since UCN-01 did not significantly affect the cytotoxicity of PMA or EtOH (Figure 2B).
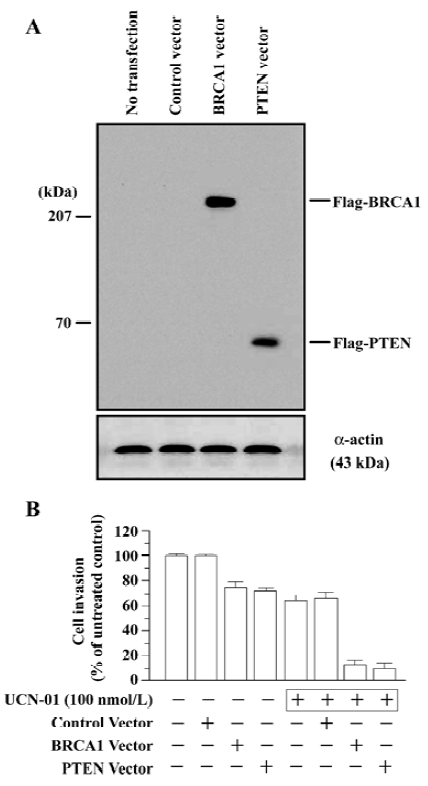
Effect of BRCA1 and PTEN on anti-invasion activity of UCN-01 To determine the effect of the tumor suppressor genes BRCA1 and PTEN on the anti-invasion potential of UCN-01, we compared the invasion behavior of U-87MG cells that had been transiently transfected with the BRCA1 or PTEN gene in the presence and absence of UCN-01 (100 nmol/L). The full-length BRCA1 or PTEN cDNA was expressed in a pCMV-Tag2B vector, which allows for the expression of proteins with an N-terminal FLAG sequence. Exponentially growing cells (3×105 cell/well) in 6-well tissue culture dishes were transfected with either the BRCA1 or PTEN vector (5 μg/well) overnight, washed, and then incubated for 48 h. Overexpression of the BRCA1 or PTEN transgene was confirmed by the determination of BRCA1 or PTEN protein using an anti-FLAG antibody (M2). Significant expression levels of the BRCA1 (220 kDa) and PTEN (67 kDa) proteins were observed in U-87MG cells 48 h after transfections (Figure 3A). The transfected cells were collected and subjected to the in vitro invasion assay. As shown in Figure 3B, the transfection with the “empty” pCMV-Tag2B control vector had no effect on UCN-01 modulation of cell invasion, whereas overexpression of the BRCA1 or PTEN transgene significantly enhanced the anti-invasion potential of UCN-01. BRCA1 or PTEN transfection alone caused a 20% reduction in cell invasion; 100 nmol/L UCN-01 inhibited 30% of cell invasion. However, UCN-01 caused an 87% and a 90% inhibition in invasion in cells transfected with BRCA1 and PTEN, respectively. These results suggest that a combination of UCN-01 and BRCA1 or PTEN transgene synergistically inhibits the invasive behavior of glioblastoma cells.
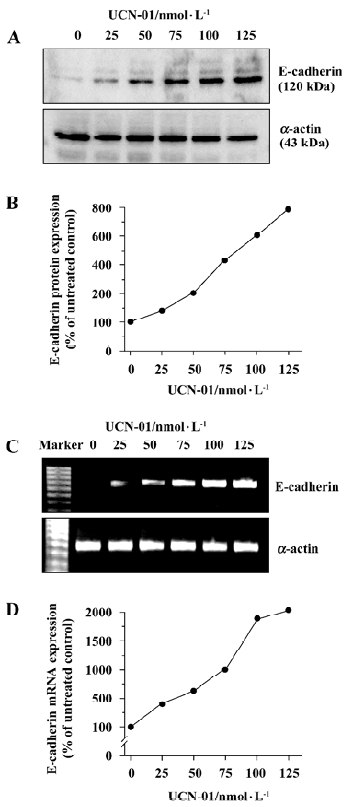
UCN-01 increases E-cadherin expression To explore the mechanism(s) underlying the effect of UCN-01 on U-87MG cell invasiveness and migration, we analyzed the expression of E-cadherin protein, an adhesion molecule, by an immunoblot assay, following treatment with UCN-01. As illustrated in Figure 4A, U-87MG cells contained a low basal level of the endogenous E-cadherin protein. Forty-eight hours exposure to UCN-01 resulted in a significant dose-dependent increase. UCN-01(50 and100 nmol/L resulted in approximate 2.2- and 6.3-fold increases in the E-cadherin protein. Similar results were obtained for E-cadherin mRNA expression in response to UNC-01 treatment (Figure 4C).
Effect of UCN-01 and anti-E-cadherin antibody on formation of cell-cell aggregates To ascertain whether E-cadherin plays an important role in the protection of UCN-01 against the migration and invasion activity of U-87MG cells, we examined the effect of UCN-01 on the formation of cell-cell aggregates. UCN-01 100 nmol/L significantly inhibited the formation of 3-dimensional cellular aggregates (aggrega-tion index=1.3, P<0.01) compared to the untreated control U-87MG cells, which showed a significant degree of cell-cell aggregation (aggregation index=0.65). However, in the presence of the E-cadherin antibody, the protective ability of UCN-01 against the formation of cell-cell aggregates was markedly reduced (aggregation index=0.8). Similar results were observed in another human glioblastoma cell line, U-373 (data not shown). These results suggest that E-cadherin can play a critical role in the anti-invasion potential of UCN-01.
Discussion
PKC is viewed as a new target for suppressing tumor cell invasion and migration, and therefore metastasis[1,2]. In the present study we found that in human gliocarcinoma U-87MG cells, UCN-01 inhibits cell proliferation in a concentration- and time-dependent fashion at higher doses (>100 nmol/L). Furthermore, at less cytotoxic doses (<100 nmol/L), UCN-01 reduces the PKC activity and suppresses the cell invasion and migration capabilities. The anti-invasion and anti-migration potential of UCN-01 was confirmed by further experiments in which the PMA- or EtOH-promoted activities of cell invasion were blocked by UCN-01. Both PMA and EtOH are potent activators of PKC[1,18]. These findings corroborate previous observations that PKC plays a central role in the invasion of glioblastoma-derived cell lines[19]. Therefore, the profound inhibition of tumor cell invasion and migration are novel features that contribute to the anti-tumor properties of UCN-01.
Although the mechanisms by which suppression of cell invasion and migration by UCN-01 have yet to be discovered, our study has revealed an important finding: despite the inhibition of PKC activity, up-regulated expression of the cell adhesion E-cadherin protein accompanies UCN-01-modulated cell invasion and migration activity. Moreover, a significant reduction of UCN-01 resulted in increased formation of cell-cell aggregates by the E-cadherin antibody. E-cad-herin, a transmembrane glycoprotein, is a key mediator of cell-cell adhesion, which acts via the formation of a complex with 3 major cytoplasmic catenins (α, β, and γ), and reportedly plays a key role in controlling the invasive and metastatic progression of a variety of human carcinoma cells, including glioblastoma[20]. Disruption of the E-cadherin/catenin complex, due primarily to decreased or lost expression of E-cadherin, is shown to be correlated with increased cell proliferation, motility, and invasiveness associated with the progression of tumors[21]. Moreover, it is also found that tamoxifen, a PKC inhibitor, blocks the invasion and migration of tumor cells by increasing the expression and functions of E-cadherin/catenin complexes[1]. Therefore, E-cadherin is believed to be a critical mediator of the anti-tumor invasion and migration potential of UCN-01, although further experiments are needed for clarification of the relationship between E-cadherin expression and PKC activity.
In addition, we also found that the enforced expression of two tumor suppressor genes, BRCA1 or PTEN, by a transient transfection assay significantly affects the inhibition potential of UCN-01 on U-87MG glioblastoma cell invasion in a synergistic manner. BRCA1 (Breast Cancer susceptibility gene 1) is located at human chromosome 17q21, and mutations of BRCA1 are known to confer an added risk for breast and ovarian cancers in women, and for prostate cancer in men[22]. Increasingly, evidence indicates that invasive breast tumors show decreased BRCA1 mRNA expression and a loss of BRCA1 immunochemical staining relative to non-invasive tumors and benign tissues[23]. A clinical observation suggests that E-cadherin expression is potentially correlated with BRCA1-associated breast cancer[24]. Our recent studies also found that indole-3carbinol, a promising phytochemical produced by cruciferous vegetables, results in up-regulation of BRCA1 and E-cadherin/catenin complex expression, which results in the suppression of tumor invasion and migration[17]. In addition, we also found that alcohol promotes tumor cell invasion and migration and is associated with down-regulation of BRCA1[24].
Phosphatase and tensin homologue deleted from chromosome 10 (PTEN) was identified in the 10q23 chromosome region and is often found in mutated forms in a wide range of human malignancies, including glioblastoma[25]. The restoration of the PTEN gene into U-87MG cells has been found to markedly suppress cell migration and invasion by negative regulation of the signals generated at the focal adhesions, and by the direct dephosphorylation and inhibition of FAK, which is involved in the cell’s interactions with extracellular matrix proteins responsible for the migration and enhancement of cell spreading through phosphorylation of its tyrosine[26]. Loss of PTEN expression and inactivation of wild-type PTEN function can often be observed in advanced or invasive tumors. Accordingly, these findings suggest that both BRCA1 and PTEN can function as inhibitors of tumor invasion and metastasis. Thus, our present findings provide in vitro evidence to suggest that a combination of UCN-01 function along with BRCA1 or PTEN gene therapy could be potentially useful in the clinical prevention and treatment of glioblastoma, although it is evident that for this to occur, further studies are required.
In conclusion, we discovered that UCN-01, as a selective PKC inhibitor, suppressed the cell migration and invasion and inhibits the PMA- and EtOH-promoted cell invasion in human glioblastoma U-87MG cells. Moreover, this inhibition of glioblastoma cell invasion and migration by UCN-01 is characterized by an enhanced expression of cell adhesion molecule E-cadherin. Therefore, UCN-01 can be a potent anti-tumor agent in the therapy of glioblastoma, not only via the suppression of proliferation of glioblastoma cells, but also via the inhibition of cell invasion and metastasis. Our results, which support previous studies[19], also indicate that the PKC signal transduction pathway can play an important role in glioblastoma cell invasion and metastasis, and that the suppression of this pathway may significantly impair the malignant progression of human glioblastoma.
References
- Caponigro F, French RC, Kaye SB. Protein kinase C: a worthwhile target for anticancer drugs? Anticancer Drugs 1997;8:26-33.
- Gomez DE, Skilton G, Alonso DF, Kazanietz MG. The role of protein kinase C and novel phorbol ester receptors in tumor cell invasion and metastasis. Oncol Rep 1999;6:1363-70.
- Akinaga S, Sugiyama K, Akiyama T. UCN-01(7-hydroxystauro-sporine) and other indolocarbazole compounds: a new generation of anti-cancer agents for the new century? Anticancer Drug Des 2000;15:43-52.
- Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O’Connor PM. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst 1996;88:956-65.
- Shao RG, Cao CX, Pommier Y. Activation of PKCalpha downstream from caspases during apoptosis induced by 7-hydroxystaurosporine or the topoisomerase inhibitors, camptothecin and etoposide, in human myeloid leukemia HL60 cells. J Biol Chem 1997;272:31321-5.
- Shao RG, Shimizu T, Pommier Y. 7-Hydroxystaurosporine (UCN-01) induces apoptosis in human colon carcinoma and leukemia cells independently of p53. Exp Cell Res 1997;234:388-97.
- Shimizu T, Cao CX, Shao RG, Pommier Y. Lamin B phosphorylation by protein kinase calpha and proteolysis during apoptosis in human leukemia HL60 cells. J Biol Chem 1998;273:8669-74.
- Koh J, Kubota T, Koyama T, Migita T, Hashimoto M, Hosoda Y, et al. Combined antitumor activity of 7-hydroxystaurosporine (UCN-01) and tamoxifen against human breast carcinoma in vitro and in vivo. Breast Cancer 2003;10:260-7.
- Koh J, Kubota T, Migita T, Abe S, Hashimoto M, Hosoda Y, et al. UCN-01 (7-hydroxystaurosporine) inhibits the growth of human breast cancer xenografts through disruption of signal transduction. Breast Cancer 2002;9:50-4.
- Bozko P, Larsen AK, Raymond E, Skladanowski A. Influence of G2 arrest on the cytotoxicity of DNA topoisomerase inhibitors toward human carcinoma cells with different p53 status. Acta Biochim Pol 2002;49:109-19.
- Jones CB, Clements MK, Redkar A, Daoud SS. UCN-01 and camptothecin induce DNA double-strand breaks in p53 mutant tumor cells, but not in normal or p53 negative epithelial cells. Int J Oncol 2000;17:1043-51.
- Yamauchi T, Keating MJ, Plunkett W. UCN-01 (7-hydroxystau-rosporine) inhibits DNA repair and increases cytotoxicity in normal lymphocytes and chronic lymphocytic leukemia lymphocytes. Mol Cancer Ther 2002;1:287-94.
- Kruger EA, Blagosklonny MV, Dixon SC, Figg WD. UCN-01, a protein kinase C inhibitor, inhibits endothelial cell proliferation and angiogenic hypoxic response. Invasion Metastasis 1998;18:209-18.
- Kruger EA, Figg WD. Protein binding alters the activity of suramin, carboxyamidotriazole, and UCN-01 in an ex vivo rat aortic ring angiogenesis assay. Clin Cancer Res 2001;7:1867-72.
- Nahta R, Hortobagyi GN, Esteva FJ. Signal transduction inhibitors in the treatment of breast cancer. Curr Med Chem Anti-Cancer Agents 2003;3:201-16.
- Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001;20:77-87.
- Meng Q, Qi M, Chen DZ, Yuan R, Goldberg ID, Rosen EM, et al. Suppression of breast cancer invasion and migration by indole-3-carbinol: associated with up-regulation of BRCA1 and E-cadherin/catenin complexes. J Mol Med 2001;78:155-65.
- Stubbs CD, Slater SJ. Ethanol and protein kinase C. Alcohol Clin Exp Res 1999;23:1552-60.
- Zhang W, Law RE, Hinton DR, Couldwell WT. Inhibition of human malignant glioma cell motility and invasion in vitro by hypericin, a potent protein kinase C inhibitor. Cancer Lett 1997;120:31-8.
- Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 1991;113:173-85.
- Meng Q, Gao B, Goldberg ID, Rosen EM, Fan S. Stimulation of invasion and migration by alcohol in breast cancer cells. Biochem Biophys Res Commun 2000;273:448-53.
- Zheng L, Li S, Boyer TG, Lee WH. Lessons learned from BRCA1 and BRCA2. Oncogene 2000;19:6159-75.
- Seery LT, Knowlden JM, Gee JM, Robertson JF, Kenny FS, Ellis IO, et al. BRCA1 expression levels predict distant metastasis of sporadic breast cancers. Int J Cancer 1999;84:258-62.
- Jacquemier J, Eisinger F, Nogues C, Sun ZZ, Guinebretiere JM, Peyrat JP, et al. Histological type and syncytial growth pattern affect E-cadherin expression in a multifactorial analysis of a combined panel of sporadic and BRCA1-associated breast cancers. Int J Cancer 1999;83:45-9.
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997;275:1943-7.
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998;280:1614-7.