
Engagement of membrane immunoglobulin enhances Id3 promoter activity in WEHI-231 B lymphoma cells
Introduction
Signalling transduction molecule(s) through B cell antigen receptor (BCR) are considered to play a crucial role in the regulation of B cell fate. The engagement of BCR leads to activation or inactivation of B cells, depending on B cell developmental stage as well as interaction with other cells including T cells expressing CD40 ligand (CD40L)[1–3]. The molecular mechanisms underlying BCR-mediated activation and/or inactivation remain largely unresolved. WEHI-231 B lymphoma cells, representing immature stage of B cells, have been widely used as an in vitro model system to analyze B cell-unresponsiveness[4–6], since costimulation with CD40L rescues anti-IgM-induced growth arrest and/or apoptosis[7,8].
The basic helix-loop-helix (bHLH) family of transcription factors including E2A play a key role in proliferation and/or differentiation in a variety of cell types[9,10]. The E protein contains a highly conserved HLH dimerization domain and a DNA-binding basic region that binds a conserved E-box. Another category of HLH protein, the inhibitor of differentiation (Id) family possess homologous HLH domain, but not DNA-binding domain[11,12]. Thus, Id proteins function as a dominant-negative (dn) regulator of transcription upon binding with other HLH proteins. The Id proteins are thought to promote cell cycle progression from G1 to S phase, while E2A proteins are thought to prevent it[9,13]. These findings indicate that E2A/Id proteins function to promote or inhibit proliferation in a context of cell types in divergent stimuli. For example, mature B cells from Id3-deficient mice failed to proliferate in response to anti-IgM[14]. Contrary to this current notion, we have recently demonstrated that anti-IgM-induced G1 arrest appears to result from the anti-IgM-induced upregulation of Id3 expression at both mRNA and protein levels in WEHI-231 cells[15], as reported in other cell types[16]. In the present study, we examined whether anti-IgM-induced upregulation of Id3 mRNA resulted from transcriptional regulation of the Id3 gene.
Materials and methods
Cell culture WEHI-231 B lymphoma cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 2-mercaptoethanol 50 µmol/L, and kanamycin 100 mg/L at 37 ºC in 5% CO2.
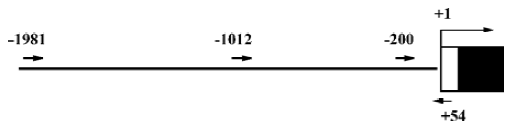
Isolation of 5'-flanking region of mouse Id3 Genomic DNA was extracted from WEHI-231 cells using QIAGEN Genomic DNA Kit according to the manufacturer’s instruc-tion. DNA fragments corresponding to the 5'-flanking region of the mouse Id3 gene were isolated from the genomic DNA by polymerase chain reaction (PCR) using Id3-specific primers and high fidelity KOD DNA polymerase (Toyobo Co, Ltd, Japan) (Figure 1), based on the reported nucleotide sequences (GenBank accession number, AL935264). The nucleotide sequences of the primers were the following: forward with a Kpn I restriction enzymatic site underlined, Id3 -1981/+54, (5'-CGGGGTACCCATACAGAAGTCTTCA-CGAGAG-3); Id3-1012/+54 (5'-CGG GGTACC TGGCTGCC-CTTCCACAACAG-3'); Id3 -200/+54 (5'-CGG GGTACC TCCTCGCATCCGAGGCTCC-3'). Reverse for the three fragments with a Hind III restriction enzymatic site underlined, (5'-CGG AAGCTT GAGAGTAGAGATAGAGAGGGAG-3'). The DNA fragments Id3 -1981/+54 (Id3/2k), Id3 -1012/+54 (Id3/1k), and Id3 -200/+54 (Id3/254) were cloned into pCR-Blunt II-TOPO vector by Zero Blunt PCR Cloning Kit (Invitrogen, USA). The nucleotide sequences of the fragments were confirmed to be identical to the reported sequences (GenBank) using ABI PRISM BigDyeTM Terminator Cycle Sequencing Ready Reaction Kits (Applied Biosystems, USA).
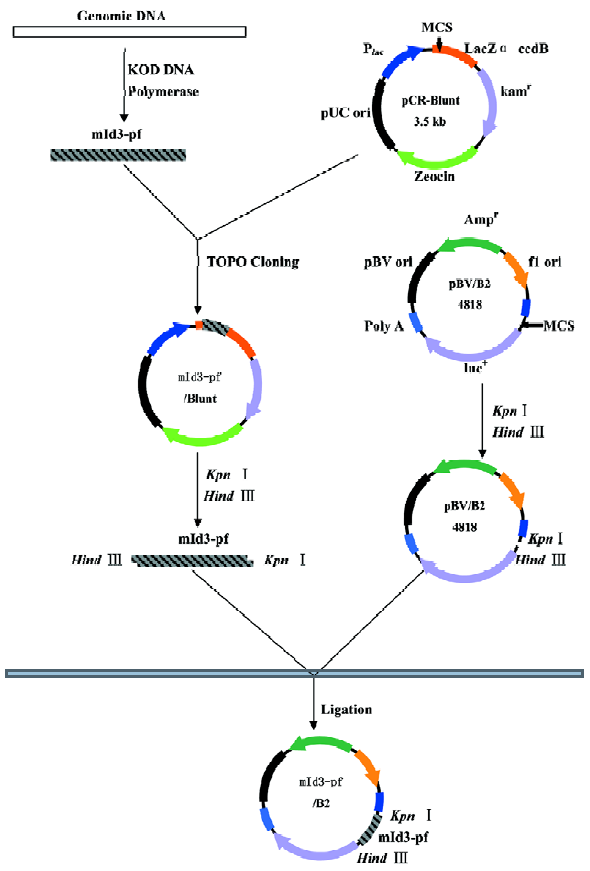
Plasmid construction for transient transfection assay The fragments of the Id3 promoter spanning nucleotides -1981/+54, -1012/+54, and -200/+54 were subcloned into the firefly luciferase reporter PGV-B2 vector lacking the promoter and enhancer (Tokyo-Inki, Tokyo, Japan) (Id3/PBV-B2), as previously described[17], resulting in Id3/2k-Luc, Id3/1k-Luc, and Id3/254-Luc, respectively.
Transient transfection of WEHI-231 cells was carried out by an electroporation method[17]. WEHI-231 cells (1.2×107 per 0.7 mL) containing 20 μg of the reporter constructs and 400 ng of internal control pRL-CMV Renilla luciferase vector were pulsed (400 V, 500 µF) using Gene Pulser (Bio-rad, USA). Following a 24-h incubation, the cells were further stimulated with 10 mg/L anti-IgM mAb or medium alone for 24 h, or cocultured with irradiated CD40L-expressing NIH3T3 cells or control NIH3T3 cells. The cells were harvested and lysed in lysis buffer (Promega, USA), followed by an assay for luciferase activity using a Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer’s instruc-tions. Both firefly and Renilla luciferase activities were monitored with a Lumat LB9507 luminometer (Bert-hold). The ratio of firefly/renila values was caculated to normalize report activity. Normalized data are expressed as relative luciferase activity compared with that seen for PGV-B2 vector alone.
Statistical analysis Data were expressed as mean±SD. For single comparison, statistical significance between groups was determined by Student’s t-test. Significance for multiple comparisons was determined by one-way ANOVA. P<0.05 was considered to indicate statistically significant differences.
Results
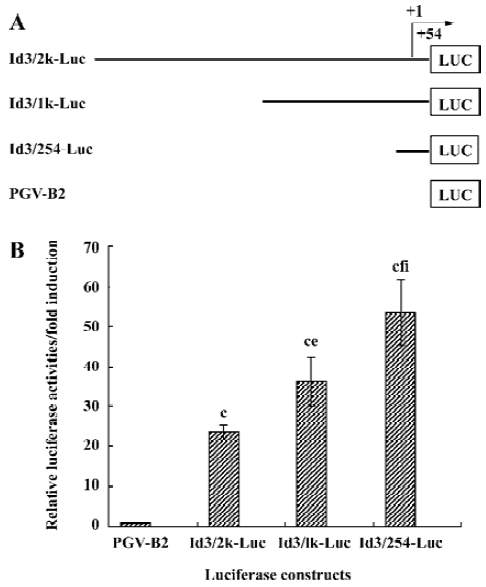
Structure of the functional Id3 locus We have recently found that anti-IgM upregulated Id3 mRNA expression, while CD40L downregulated it in WEHI-231 cells[15]. To analyze whether engagement of mIg influences transcriptional regulation of the Id3 gene, the three fragments containing the 5'-flanking region of the murine Id3 gene were isolated by PCR using Id3-specific primers. These fragments were subcloned into PGV-B2 vector to measure the promoter activity of the Id3 gene (Figure 2). Nucleotide sequence analysis confirmed the fidelity of the all constructs generated.
Basal promoter activity in the 5'-flanking region of the Id3 gene WEHI-231 cells were transfected with the constructs Id3/2k-Luc, Id3/1k-Luc, or Id3/254-Luc in combination with internal control vector and incubated with medium alone for 24 h, followed by assay for luciferase activity (Figure 3A, 3B). The basal promoter activity in these fragments Id3/2k, Id3/1k, or Id3/254 was increased 24-fold, 36-fold, and 54-fold, respectively, compared with the control vector (Figure 3B). These results suggest that the 254-bp fragment contains some positive regulatory element(s) of the Id3 gene, as reported by Yeh and Lim[18]. It is also possible that there are putative negative regulatory elements in the region from -1981 to -201.
Anti-IgM, but not CD40L, upregulates the promoter activity of the Id3 gene To examine whether transcriptional regulation is involved in the anti-IgM-induced upregulation of the Id3 gene, the WEHI-231 cells co-transfected with the Id3/2k-Luc, Id3/1k-Luc, Id3/254-Luc constructs and internal control vector were stimulated with 10 mg/L anti-IgM for 24 h, followed by assay for luciferase activity. The stimulation with anti-IgM enhanced the promoter activity of the Id3 gene which increased up to two-fold as compared with the activities in unstimulated cells, as assessed by luciferase activity (Figure 4A). The CD40L stimulation slightly decreased it, but the difference was not statistically significant (Figure 4B).
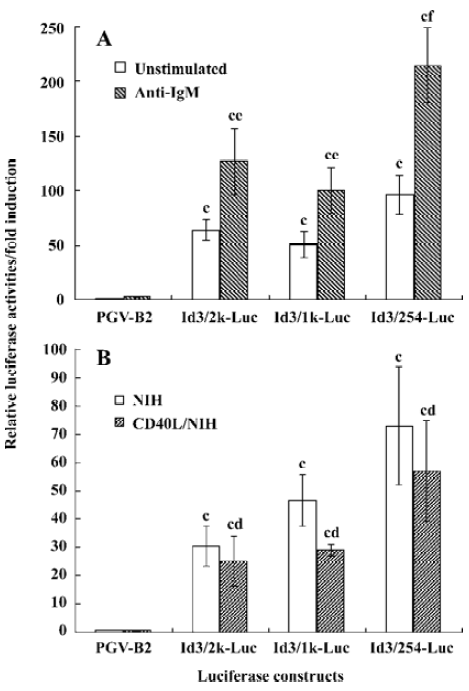
In our previous study, with Northern blot assay we observed an upregulation of Id3 mRNA expression in WEHI-231 cells after anti-IgM stimulation to reach levels 5-fold greater than that seen in unstimulated cells. There are many reasons to explain the inconsistent results between luciferase activity assay and northern blot. Besides the stability of the luciferase and mRNA, additional mechanisms may exist that contribute to the upregulation of Id3 expression at transcription level following anti-IgM stimulation. These results suggest that the mIg-mediated upregulation of Id3 expression is controlled, at least in part, through transcriptional regulation, as assessed by luciferase assay.
Discussion
Id proteins, a member of the HLH family proteins, play a crucial role in proliferation, differentiation, and apoptosis in a variety of cells including lymphocytes[9–11]. The mature B cells from Id3-deficient mice failed to proliferate in response to anti-IgM, whereas they showed proliferation comparable to those from control mice in response to LPS or CD40L[14], suggesting that Id3 promotes mIg-mediated B cell growth, as demonstrated in some cell types[11]. However, WEHI-231 cells, representing immature B cells, displayed growth arrest at the G1 phase of the cell cycle, accompanied by upregula-tion of Id3 mRNA following stimulation with anti-IgM[15]. In this study we examined whether the mIg-mediated upregula-tion of Id3 mRNA expression resulted from transcriptional activation and found that anti-IgM upregulated Id3 expression at least through transcriptional activation.
The nucleotide sequences of the 5'-upstream fragments isolated from WEHI-231 genomic DNA were confirmed to be identical with the published sequences[18] (GenBank accession number, AL935264). Consistent with the observation of Yeh and Lim[18] using C2C12 muscle cells, the 254-bp fragment had substantial basal promoter activity of the Id3 gene (Figure 3). Indeed, several putative binding sites (ATF, Sp1, Egr1, Cre) for transcription factors were found using TRANSFAC databases[18]. Interestingly, when the longer fragments Id3/2k-Luc or Id3/1k-Luc was used, the basal promoter activity decreased compared with Id3/254-Luc (Figure 3B). These results might suggest that the promoter activity of the Id3 gene be regulated by both positive and negative regulatory elements, as reported in the transcriptional regulation of a variety of genes in eukaryotes[19]. A sufficient promoter activity was also found in other several B lymphoid lineage cell lines BAL17 and J558L (Li and Hata unpublished observation, 2003), although it is not dependent on B cell development.
The promoter activity of the Id3 gene was increased following engagement of mIg, but not CD40 (Figures 4A and 4B), resulting in the upregulation of the Id3 mRNA in WEHI-231 B lymphoma cells[15]. Although the Id3 upregulation through engagement of T cell receptor for antigen was abrogated by MEK1 inhibitor PD98059[20], the mIg-mediated upregulation of the Id3 message was not affected (Li and Hata, unpublished observation), suggesting that antigen receptor-mediated upregulation of Id3 expression is differently regulated between T and B cells.
In contrast to anti-IgM, CD40L decreased Id3 mRNA levels in WEHI-231 cells[15]. The promoter activity of the Id3 gene in response to CD40L was somewhat low, but not significant, compared with medium alone, suggesting that CD40-mediated down-regulation of Id3 mRNA expression is controlled post-transcriptionally. In our preliminary experiments, pretreatment with proteasomal inhibitor lactacystin restored CD40L-mediated down-regulation of Id3 protein expression (Li and Hata, unpublished observation), as suggested by Bounpheng et al[21]. These findings suggest that CD40-mediated down-regulation is controlled post-translationally.
Following an encounter with antigen, immature B cells undergo growth arrest/apoptosis unless T cell-derived signals such as CD40L are provided[7,22]. Since transduction of Id3 gene into WEHI-231 cells results in growth arrest at the G1 phase of the cell cycle[15], Id3 appears to contribute to G1 arrest in immature B cells. The cell cycle progression from G1 to S phase in mammalian cells is tightly regulated by a cyclin-dependent kinase (CDK) and cyclin[23]. CDK activity is controlled by CDK inhibitors (CKIs) including p21Cip1 and p27Kip1[24]. Anti-IgM induced growth arrest through an upregulation of p27Kip1 and down-regulation of cyclins[25]. It remains unclear how Id3 relates to p27Kip1-mediated signalling pathway in B cells.
In contrast to our findings, Pan et al reported that anti-IgM-induced proliferative responses were severely reduced in mature B cells from Id3-deficient mice, compared with those from control mice[14]. Id3 appears to promote cell cycle progression in mature B cells upon encounter with antigen, whereas it prevents cell cycle progression in immature B cells, which could be accounted for by differential target(s) of Id3 molecule between mature and immature B cells. Experiments to examine this possibility are in progress.
In the present study, we clarified that engagement of mIg increased the promoter activity of the Id3 gene, probably resulting in an enhanced Id3 expression. The mIg-mediated Id3 upregulation appears to contribute to growth arrest, which could be reversed by the addition of CD40L, at least in part, through down-regulation of Id3 expression. Our results would be valuable for analysis of mIg-mediated unresponsiveness in B cells.
References
- Nossal GJ. Immunologic tolerance: collaboration between antigen and lymphokines. Science 1989;245:147-53.
- Klaus GG, Choi MS, Lam EW, Johnson-Leger C, Cliff J. CD40: a pivotal receptor in the determination of life/death decisions in B lymphocytes. Int Rev Immunol 1997;15:5-31.
- DeFranco AL. The complexity of signaling pathways activated by the BCR. Curr Opin Immunol 1997;9:296-308.
- Donjerkovic D, Scott DW. Activation-induced cell death in B lymphocytes. Cell Res 2000;10:179-92.
- Benhamou LE, Cazenave PA, Sarthou P. Anti-immunoglobulins induce death by apoptosis in WEHI-231 B lymphoma cells. Eur J Immunol 1990;20:1405-7.
- Scott DW, Livnat D, Pennell CA, Keng P. Lymphoma models for B cell activation and tolerance. III. Cell cycle dependence for negative signalling of WEHI-231 B lymphoma cells by anti-mu. J Exp Med 1986;164:156-64.
- Scott DW, Chace JH, Warner GL, O'Garra A, Klaus GG, Quill H. Role of T cell-derived lymphokines in two models of B-cell tolerance. Immunol Rev 1987;99:153-71.
- Tsubata T, Wu J, Honjo T. B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature 1993;364:645-8.
- Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 2000;20:429-40.
- Engel I, Murre C. The function of E- and Id proteins in lymphocyte development. Nat Rev Immunol 2001;1:193-9.
- Norton JD, Deed RW, Craggs G, Sablitzky F. Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol 1998;8:58-65.
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 1990;61:49-59.
- Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci 2000;113:3897-905.
- Pan L, Sato S, Frederick JP, Sun XH, Zhuang Y. Impaired immune responses and B-cell proliferation in mice lacking the Id3 gene. Mol Cell Biol 1999;19:5969-80.
- Hata K, Yoshimoto T, Mizuguchi J. CD40 ligand rescues inhibitor of differentiation 3-mediated G1 arrest induced by anti-IgM in WEHI-231 B lymphoma cells. J Immunol 2004;173:2453-61.
- Zhao F, Vilardi A, Neely RJ, Choi JK. Promotion of cell cycle progression by basic helix-loop-helix E2A. Mol Cell Biol 2001;21:6346-57.
- Hata K, Mizuguchi J. Genomic organization and characterization of the promoter for the E2A gene. Gene 2004;325:53-61.
- Yeh K, Lim RW. Genomic organization and promoter analysis of the murine Id3 gene. Gene 2000;254:163-71.
- Ogbourne S, Antalis TM. Transcriptional control and the role of silencers in transcriptional regulation in eukaryotes. Biochem J 1998;331:1-14.
- Bain G, Cravatt CB, Loomans C, Alberola-Ila J, Hedrick SM, Murre C. Regulation of the helix-loop-helix proteins, E2A and Id3, by the Ras-ERK MAPK cascade. Nat Immunol 2001;2:165-71.
- Bounpheng MA, Dimas JJ, Dodds SG, Christy BA. Degradation of Id proteins by the ubiquitin-proteasome pathway. FASEB J 1999;13:2257-64.
- DeFranco AL, Gold MR, Jakway JP. B-lymphocyte signal transduction in response to anti-immunoglobulin and bacterial lipopolysac-charide. Immunol Rev 1987;95:161-76.
- Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997;13:261-91.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999;13:1501-12.
- Ezhevsky SA, Toyoshima H, Hunter T, Scott DW. Role of cyclin A and p27 in anti-IgM induced G1 growth arrest of murine B-cell lymphomas. Mol Biol Cell 1996;7:553-64.