
Novel role for gabapentin in neuroprotection of central nervous system in streptozotocine-induced diabetic rats1
Introduction
Diabetes mellitus is a common metabolic disorder that affects the peripheral as well as the central nervous system. Animal models of diabetes can make an important contribution to understanding the pathology of the effects of diabetes on the brain. Hyperglycemia provides more substrate for anaerobic glycolysis in the brain, resulting in lactic acidosis and enhanced damage to both glial and neuronal cells[1]. Furthermore, enhanced formation of oxygen free radicals occurs in the tissues during hyperglycemia[2]. These oxidant radicals also contribute to increased neuronal death by oxidizing proteins damaging DNA and inducing the lipoperoxidation of cellular membranes[3].
One of the complications of diabetes is neuropathy. Gabapentin (GBP) is used to treat diabetic neuropathy. GBP is an anticonvulsant that reduces pain in peripheral neuro-pathy, including diabetic neuropathy[4]. GBP is a hydrophilic analogue of the inhibitory neurotransmitter gamma aminobutyric acid (GABA). It readily penetrates the blood-brain barrier via an L-system amino acid transporter system[5]. In addition, GBP has been shown to be neuroprotective in animals[6]. However, to date, no study has examined the effects of GBP on neural and glial markers.
S100B and neuron-specific enolase protein (NSE), distributed mainly in the central nervous system, can provide qualitative information about the extent of brain injury and are sensitive markers of brain damage after stroke and cerebral hypoxia[7,8]. NSE is a glycolytic enzyme that is a soluble cytoplasmatic protein localized principally in neurons. Recent evidence has shown that NSE is a marker of neuronal damage following cerebral ischemia[8,9]. S100B is an acidic Ca2+ binding protein, present mainly in astrocytes, that exerts paracrin trophic effects on several neuronal populations[10]. Brain injury causes a selective leakage of S100B into the cerebrospinal fluid and blood so that serum S100B levels were found to be good indicators for the assessment of patients with cerebral ischemia[11,12].
Glial fibrillary acidic protein (GFAP) is another astrocytic marker. GFAP is an intracellular intermediate filament protein of glial cells. It has been suggested that GFAP is essential for the formation of stable astrocytic processes in response to neuronal damage and this may be critical for morphogenesis of the central nervous system[13]. Chemical or mechanical insults to the brain cause permanent changes, with astrocytes responding via a variety of reactions called reactive gliosis[14]. A key indicator of glial reactivity is the increased synthesis of GFAP. Increases in GFAP are commonly used to examine the distributions of glial cells in response to neural injury[14,15]. Recently, we have demonstrated that streptozotocine (STZ)-induced diabetes causes reactive gliosis[15,16]. In the present study we aimed to examine the effects of GBP on neural and glial markers in STZ-induced diabetic rats.
Materials and methods
Sixty adult male Wistar rats (Animal Research Unit, Firat University, Elazig, Turkey) weighing 200–250 g were used in this study. The rats were housed in a temperature-controlled room (22–25 ºC) with a 12/12 h light/dark cycle. Water and food were given ad libitum. Animals were assigned into a diabetic group (n=30) or an age-matched control group (n=30). Diabetes was induced by a single intraperitoneal (ip) injection of 50 mg/kg body weight STZ. STZ (Sigma Chemical Company, St Louis, MO, USA) was dissolved in sodium citrate buffer (pH 4.5). Blood glucose concentrations were determined 3 d after STZ injection. Rats with a blood glucose concentration above 250 mg/dL were declared diabetic. Diabetic rats were randomly assigned to two groups: the first group received GBP (Sigma) diluted in sterile physiological saline and administered ip (50 mg·kg-1·d-1) in a volume of 1 mL/kg. (STZ-GBP group, n=15). The second group received the vehicle alone (STZ group; n=15). Control rats were injected with the vehicle alone (n= 15) or with the same dose of GBP (GBP group; n=15). Each animal’s body weight and diabetic state were reassessed after 6 weeks just prior to killing the animals. All animals were killed by decapitation after 5 weeks of treatment by GBP. The brain tissues were removed; the hippocampus, cortex, and cerebellum were dissected. Samples were used fresh or kept at -70 ºC. All protocols described were reviewed and approved by the local Institutional Committee for the Ethical Use of Animals.
Western blotting Tissue samples were homogenized in Tris-HCl 10 mmol/L (pH 7.4), NaCl 0.1 mmol/L, phenylmethyl-sulphonyl fluoride 0.2 mmol/L, edetic acid 5 mmol/L, β-mercaptoethanol 2 mmol/L, and 1% Triton X-100 containing proteinaze inhibitors, and centrifuged at 40 000×g at 4 ºC for 60 min. Supernatants were collected, aliquoted, and stored at -70 ºC until required.
Sodium dodecyl sulfate (SDS)-polyacrylamide gradient gel electrophoresis was carried out as previously described[17]. Samples and standard protein markers were submitted to the SDS-polyacrylamide gradient gel and the separated proteins were transferred to nitrocellulose filters (Schleicher & Schuell Incorporated, Keene, NH, USA). Non-specific binding was blocked by incubation with 1% bovine serum albumin. The blots were then incubated with primary antibodies (Santa Cruz Biotechnology Incorporated, Santa Cruz, CA, USA) as follows: GFAP 1:2 000; S100 1:1000; NSE 1:1000. After a 1-h incubation the blots were washed extensively in TBS-Tween (Tris-HCl 25 mmol/L, NaCl 0.2 mmol/L, 0.1% Tween-20). The blots were then incubated for 1 h with a secondary antibody, a goat anti-rabbit Ig peroxidase conjugated (Sigma, Dorset, UK). Specific binding was detected using diaminobenzidine and H2O2 as the substrates. The relative amounts of immunoreactive GFAP, S100B, and NSE were quantified in arbitrary units by scanning blots using a computerized software program (LabWorks 4.0; UVP Incorporated, Cambridge, UK).
Levels of tissue protein, glutathione, and LPO Protein determinations were carried out according to the Lowry procedure using a protein assay kit (Sigma Chemical Company, Deisenhofen, Germany). Tissue lipid peroxidation (malondi-aldehyde+4-hydroxyalkenals) was determined using a LPO-586 kit (Oxis International Incorporated, Corvallis, OR, USA); the method is based on the reaction of N-methyl-2-phenyl-indole with malondialdehyde and 4-hydroxyalkenals at 45 °C. Glutathione (GSH) levels were determined using the method of Ellman[18]. Tissue homogenates were centrifuged with 5% trichloroacetic acid to centrifuge out the proteins. Two milliliters of phosphate buffer, 0.5 mL of 5,5' dithiobis (2-nitrobenzoic acid) and 0.4 mL of distilled water was added to 0.1 mL of this homogenate. The mixture was vortexed and the absorbance was read at 412 nm.
Statistical analysis Results were expressed as mean±SD and significant differences between groups were evaluated using ANVOA followed by a post-hoc Bonferroni test. A level of significance of P<0.05 was considered to be statistically significant.
Results
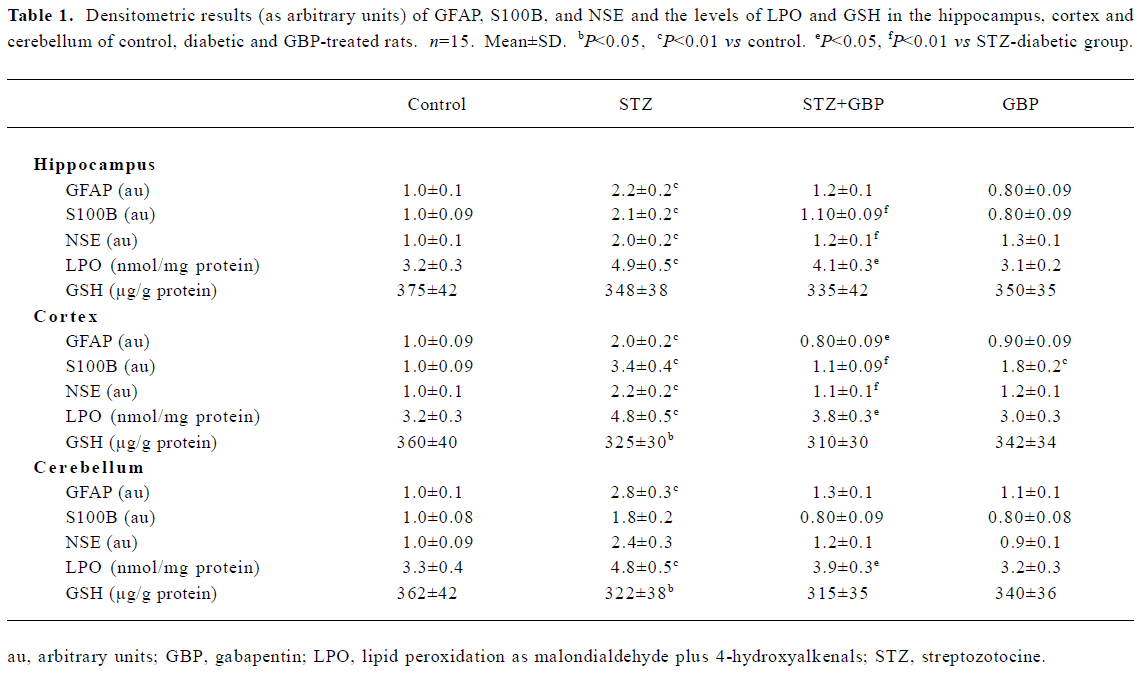
Estimation of glial and neuronal markers A common marker for reactive gliosis is an increase in GFAP. GFAP showed many immunopositive protein bands with relative molecular weights of 49 kDa (main band) and several small molecular weight bands at roughly 42–47 kDa (degraded product of GFAP), respectively (Figure 1). Although Western blot measurement of brain 49 kDa GFAP contents after 6 weeks of STZ-induced diabetes showed no significant difference, the amount of degraded GFAP product increased significantly compared with the non-diabetic controls. The beneficial effects of the repeated administration of GBP were manifested by downregulation of the degradation products of GFAP (Figure 1, Table 1).
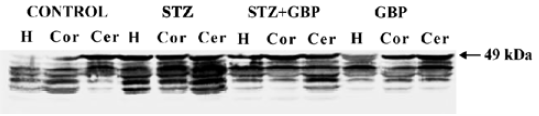
Full table
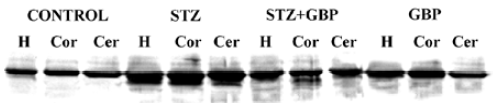
The immunoblots of the brain homogenates showed a single S100B-immunoreactive polypeptide band with a molecular weight of approximately 11 kDa. The levels of S100B protein in the hippocampus, cortex, and cerebellum were significantly higher in STZ-treated rats than that in the control group. Similarly, GBP prevented the increase of S100B protein in all studied brain areas (Figure 2, Table 1).
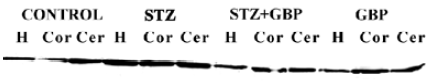
In the brain regions of STZ-treated rats, the NSE contents showed a significant increase compared with the controls. Similarly to GFAP and S100B, NSE levels were reduced by treatment with GBP (Figure 3, Table 1).
Estimation of LPO and GSH Likewise, STZ-induced diabetes led to a marked elevation in the levels of malondial-dehyde+4-hydroxyalkenals in the cortex, hippocampus, and cerebellum compared with the control group. A significant positive correlation was found between the levels of malondi-aldehyde +4-hydroxyalkenals and the GFAP contents in the cortex and hippocampus (r=0.625, P<0.01; r=0.604, P<0.01, respectively). Treatment with GBP significantly reduced the levels of malondialdehyde+4-hydroxyalkenals in the brain homogenates of diabetic rats compared to the controls (Table 1).
In the present study we measured the amount of GSH in the homogenates of cortex, hippocampus and cerebellum. In each of these brain parts, the levels of GSH were lower in the STZ-diabetic rats than in the control animals. Treatment with GBP led to a slight but non-significant reduction in the levels of GSH compared with the levels in the STZ group (Table 1).
Discussion
In the experiment reported here, we investigated the effects of GBP on glial and neuronal markers in different brain regions of diabetic rats. We found that diabetes induced overexpression in both glial and neuronal markers and GBP prevented the elevation of these markers. Interestingly, treatment with GBP did not change the levels of both neuronal and glial markers in non-diabetic rats. Furthermore, GBP treatment moderately inhibited, but did not completely reverse, the elevation in LPO induced by STZ diabetes. GFAP and S100B can be trophic or toxic depending on their concentrations. Over production of both glial markers causes glial and neuronal death, whereas in low concentrations they can act as neurotrophic factors, which may be important during both development and nerve regeneration[19,20].
Herein we report a new role for GBP that is associated with neuroprotection. These data provide the first indication that GBP regulates the expression of neural and glial markers. Although the mechanism of action of GBP was not specifically investigated in this study, the present findings provide some clues.
It has been reported that GBP is neuroprotective in chronic neural diseases[21]. Current findings appear to support this idea. The mechanism by which GBP reduced LPO levels and inhibited the expression of neuronal and glial markers is not known. This process may occur through a free radical mediated pathway. Tissue damage is observed during untreated diabetes with very high glucose levels and this is attributed to oxidative injury resulting from the production of free radicals[2,22]. One of the consequences of such damage is the depletion of the cellular antioxidant, GSH. A decreased defense system against free radicals and elevated glutamatergic activity in diabetes may cause glial sensitivity[23]. Diabetes-related sensitivity to oxidative stress of astrocytes makes neurons more susceptible to injury. Neuronal and glial injury leads to the over production of NSE, GFAP, and S100B. In agreement with the present findings we recently hypothesized that glial reactivity was closely related to the oxidative environment[15].
The current results show that treatment with GBP significantly prevents glial reactivity by reducing the over production of GFAP and S100B. Similarly GBP inhibited the elevation of NSE, a neuronal marker. The exact mechanism by which GBP inhibits reactive gliosis is not entirely clear; however, one possible explanation is that GBP may reduce oxidative stress via an unknown mechanism. Oxidative stress is known to exacerbate glial reactivity[15,16]. Decreased LPO levels as a result of GBP treatment in the present study supports this explanation.
Hyperglycemia causes a reduction in extracellular GABA, which exacerbates neuronal injury by reducing generalized neuroinhibition[23,24]. Reduced GABAergic action in the cerebral cortex in diabetes has been shown to be increased by GBP treatment[25]. Several investigations have confirmed that GBP has the ability to increase the GABA content of the brain, influence GABA receptor activity, and exhibit anticonvulsant activity[26,27]. An increase in GABAergic activity by GBP would promote inhibitory neurotransmission in the central nervous system. This is a potent mechanism by which GBP acts as a neuroprotector.
The other possible explanation for the effect of GBP is a drug-induced decrease in glutamatergic neurotransmission[28]. Glutamate neurotoxicity has been implicated in the neuro-degeneration associated with several neurodegene-rative diseases. Diabetes may increase the release of glutamate and activate NMDA receptors[29]. Glutamate causes a transmembrane ion imbalance, in particular a calcium influx, which in turn generates reactive oxygen species. These free radicals actively attack macromolecules within neuron and glial cells, resulting in structural and functional changes in proteins. Glial cells respond to the oxidative insult by producing GFAP and S100B[30,31]. The present findings are in agreement with recent reports that clearly show that reactive gliosis occurs in diabetes, possibly as a result of oxidative stress. This is consistent with our previous studies in diabetic rats in which the protective effects of antioxidants against reactive gliosis were attributed to their free radical scavenging properties[15,16]. As we previously described, antioxidant vitamin E[32] and melatonin[15] treatment improved reactive gliosis induced by hyperglycemia via a dramatic reduction in oxidative stress. Compared with antioxidants, the effect of GBP on oxidative stress is considerably more modest. The present results suggest that GBP prevents reactive gliosis and oxidative stress in an indirect way, possibly by reducing glutamate neurotoxicity, which causes free radical production in brain tissue. Previous studies have shown that repeated administration of GBP inhibits excitatory glutamatergic neurotransmission via direct blockage of excitatory receptors or alteration of glutamate metabolism or release[32]. In the present study we have shown that the production of glial and neuronal markers is increased in the brain by hyperglycemia and that this increase is attenuated by treatment with GBP. Thus, we predict that GBP treatment will attenuate the hippocampal and cortical neurodegenera-tion observed during diabetes mellitus in rats.
References
- Biessels GJ, Kappelle AC, Bravenboer B, Erkelens DW, Gispen WH. Cerebral function in diabetes mellitus. Diabetologia 1994;37:643-50.
- Baydas G, Canatan H, Turkoglu A. Comparative analysis of the protective effects of melatonin and vitamin E on streptozocin-induced diabetes mellitus. J Pineal Res 2002;32:225-30.
- Hawkins CL, Davies MJ. Generation and propagation of radical reactions on proteins. Biochim Biophys Acta 2001;1504:196-219.
- Field MJ, McCleary S, Hughes J, Singh L. Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain 1999;80:391-8.
- Su TZ, Lunney E, Campbell G, Oxender DL. Transport of gabapentin, a gamma-amino acid drug, by system l alpha-amino acid transporters: a comparative study in astrocytes, synapto-somes, and CHO cells. J Neurochem 1995;64:2125-31.
- Lagreze WA, Müler-Valten R, Feuerstein TJ. The neuroprotective properties of gabapentin-lactam. Graef’s Arch Clin Exp Ophthalmol 2001;239:845-9.
- Buttner T, Lack B, Jager M, Wunsche W, Kuhn W, Muller T, et al. Serum levels of neuron-specific enolase and S-100 protein after single tonic-clonic seizures. J Neurol 1999;246:459-61.
- Hatfield RH, McKernan RM. CSF neuron-specific enolase as a quantitative marker of neuronal damage in a rat stroke model. Brain Res 1992;577:249-52.
- Persson L, Hardemark HG, Gustafsson J, Rundstrom G, Mendel-Hartvig I, Esscher T, et al. S-100 protein and neuron-specific enolase in cerebrospinal fluid and serum: markers of cell damage in human central nervous system. Stroke 1987;18:911-8.
- Kligman D, Marshak DR. Purification and characterisation of a neurite extension factor from bovine brain. Proc Natl Acad Sci USA 1985;82:7136-9.
- Heizmann CW, Fritz G, Schafer BW. S-100 proteins: structure, functions and pathology. Front Biosci 2002;7:1356-67.
- Misler U, Wiesmann M, Friedrich C, Kaps M. S-100 protein and neuron-spesific enolase concentrations in blood as indicators of infarction volume and prognosis in acute ischemic stroke. Stroke 1997;28:1956-60.
- Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, et al. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelina-tion. Neuron 1996;17:607-15.
- Kunkler PE, Kraig RP. Reactive astrocytosis from excitotoxic injury in hippocampal organ culture parallels that seen in vivo. J Cereb Blood Flow Metab 1997;17:26-43.
- Baydas G, Reiter RJ, Yasar A, Tuzcu M, Akdemir I, Nedzvetskii VS. Melatonin reduces glial reactivity in the hippocampus, cortex, and cerebellum of streptozotocin-induced diabetic rats. Free Radic Biol Med 2003;35:797-804.
- Baydas G, Reiter RJ, Nedzvetskii VS, Yasar A, Tuzcu M, Ozveren F, et al. Melatonin protects the central nervous system of rats against toluene-containing thinner intoxication by reducing reactive gliosis. Toxicol Lett 2003;137:169-74.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680-5.
- Ellman GL. Tissue sulphydryl groups. Arch Biochem Biophys 1959;82:70-7.
- Fano G, Mariggio MA, Angelella P, Nicoletti I, Antonica A, Fulle S, et al. The S-100 protein causes an increase of intracellular calcium and death of PC12 cells. Neuroscience 1993;53:919-25.
- Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 2001;33:637-68.
- Rothstein JD, Kuncl RW. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J Neuro-chem 1995;65:643-51.
- Ercel E, Baydas G, Akyol A, Eksioglu E, Canpolat L. The effect of vitamin E on the sciatic nerve lipid peroxidation in streptozo-tocin induced diabetes mellitus. Biomed Res 1999;10:95-101.
- Guyot LL, Diaz FG, O’Regan MH, Song D, Phillis JW. The effect of topical insulin on the release of excitotoxic and other amino acids from the rat cerebral cortex during streptozotocin-induced hyperglycemic ischemia. Brain Res 2000;872:29-36.
- Malcangio M, Tomlinson DR. A pharmacologic analysis of mechanical hyperalgesia in streptozotocin/diabetic rats. Pain 1998;76:151-7.
- Petroff OA, Rothman DL, Behor KL, Lamoureux D, Mattson RH. The effect of gabapentin on brain gamma-aminobutyric acid in patients with epilepsy. Ann Neurol 1996;39:95-9.
- Cutrer FM. Antiepileptic drugs: how they work in headache. Headache 2001;41:S3-10.
- Petroff OA, Hyder F, Rothman DL, Mattson RH. Effects of gabapentin on brain GABA, homocarnosine, and pyrrolidinone in epilepsy patients. Epilepsia 2000;41:675-80.
- Shimoyama M, Shimoyama N, Hori Y. Gabapentin affects glutamatergic excitatory neurotransmission in the rat dorsal horn. Pain 2000;85:405-14.
- Gupta M, Singh J, Sood S, Arora B. Mechanism of antinociceptive effect of nimodipine in experimental diabetic neuropathic pain. Methods Find Exp Clin Pharmacol 2003;25:49-52.
- Kaneko K, Nakamura A, Yoshida K, Kametani F, Higuchi K, Ikeda S. Glial fibrillary acidic protein is greatly modified by oxidative stress in aceruloplasminemia brain. Free Radic Res 2002;36:303-6.
- Baydas G, Nedzvetskii VS, Tuzcu M, Yasar A, Kirichenko SV. Increase of glial fibrillary acidic protein and S-100B in hippocampus and cortex of diabetic rats: effects of vitamin E. Eur J Pharmacol 2003;462:67-71.
- Fink K, Dooley DJ, Meder WP, Suman-Chauhan N, Duffy S, Clusmann H, et al. Inhibition of neuronal Ca2+ influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology 2002;42:229-36.