
Effects of spider Macrothele raven venom on cell proliferation and cytotoxicity in HeLa cells1
Introduction
Spider venoms are biochemically and biologically well studied world-wide and we know as much about spider venoms as we know about other animal venoms, such as snake venoms, bee venoms, and scorpion venoms. In general, the main toxin components of most spider venoms have been well identified as a complex mixture of proteins, polypep-tides, polyamine neurotoxins, nucleic acids, free amino acids, monoamines, and inorganic salts that cause a wide range of effects in both vertebrates and invertebrates[1–3].
As far as the pharmacology and biochemistry of spider venom is concerned, spider venom represents a wide variety of ion channel toxins, novel non-neurotoxins, enzymes and low molecular weight compounds[4].
A number of studies have examined the anti-tumor substances found in snake venom[5–9], bee venom[10–12], and scorpion venom[13]. Many toxins isolated from spider venom have been invaluable in helping to determine the role and diversity of neuronal ion channels and the process of exocytosis[14–16]. Six peptide toxins (Magi 1–6) were isolated from the Hexathelidae spider, Macrothele gigas, dealing with sodium channel (Magi 1–5) and high toxicity in lepidoptera larvae of 3.1 nmol/g (Magi 6)[17]. Toxicological assays showed diverse lethal or paralytic activities of Magi 7–16 in mice and/or insects[18]. The spider Macrothele raveni was indentified as a new species in the genus Macrothele[19]. There have been two papers published about raven toxin tests in mice in which the toxins acted as a neurotoxic peptide and blocked neuromuscular transmission[20,21]. To date there has been no study examining the venom of the spider Macrothele raveni. In the present study, the effects of M raveni spider venom on HeLa cells and possible mechanisms of action were evaluated.
Materials and methods
Reagents RPMI-1640 (Gibco Laboratories, Grand Island, NY, USA) dissolved in double distilled water with the pH value adjusted to 7.0 using NaHCO3, was disinfected and stored at - 20 ºC. Fetal calf serum was purchased from Sijiqing Biological Engineering Company (Hangzhou, China), sterilized, and stored at -20 ºC. [3H]TdR was purchased from the Atomic Energy Institute of China (Beijing, China). Verapamil, streptomycin, penicillin and 0.5% hydrocortisone were purchased from North China Pharmaceutical Company (Shijiazhuang, China). Propidium iodide (PI), bovine serum albumin (BSA), and Triton X-100 were purchased from Sigma Company (St Louis, MO, USA). The rabbit polyclonal antibody against caspase-3 and rodamine-labeled secondary antibody were purchased from Santa Cruz Biotech (Santa Cruz, CA, USA).
Spider venom Pure spider Macrothele raveni venom was collected by electrical stimulation of 15 spiders M raveni (the weight of each spider was approximately 60 g)[22]. Spider venom was dissolved in phosphate buffered saline (PBS) and centrifuged at 8000×g for 10 min to remove insoluble materials. The concentration of spider venom was adjusted to 0 (control group), 10, 20, 40 mg/L. The spider venom was freeze-dried and stored at -80 ºC until required.
Cell culture The human cervical carcinoma cell line HeLa was obtained from the Cellular Biology Institute of the Chinese Academy of Sciences (Shanghai, China). The frozen cells were defrosted, transferred into the culture medium RPMI-1640 supplemented with 10% fetal calf serum, 100 kU/L streptomycin, and 100 kU/L benzyl penicillin. The cells were grown at 37 °C under a humidified atmosphere of 5% CO2 .
Determination of apoptosis Morphological evidence of apoptosis was obtained using acridine orange-ethidium bromide (AO/EB) staining. Monolayer cell cultures in 96-well plates were used for these studies. After removal of the incubation medium, the cells were rinsed and treated with a solution composed of AO/EB (100 mg/L PBS of each dye). Cells were examined using fluorescence microscopy and photographed (Olympus, Tokyo, Japan). Viable cells were colored green with intact nuclei. Non-viable cells had bright orange chromatin. Apoptosis was demonstrated by the appearance of cell shrinkage with condensation and fragmentation of nuclei. Apoptotic cells were easily distinguished from necrotic cells because the latter appeared orange with a normal nuclear structure. This procedure was used to quantify the number of apoptotic cells after treatment with spider venom. Apoptosis was also quantified using an enzyme-linked immunosorbent assay (ELISA) kit (Bindazyme; the Binding Site, Birmingham, UK) that measured the amounts of mono- and oligo-nucleosomes produced in the cytoplasmic fraction of lysed cells as a consequence of DNA fragmentation[23]. After treatment with spider venom, cells were collected and lysed according to the manufacturer’s instructions. After centrifugation at 11 000×g for 15 min, equal amounts (0.3 µg of protein) of the cytosolic fractions were used for the assays.
Specimens for electron microscopy HeLa cells in the exponential phase were used and cultivated with various concentrations of spider venom for 24 h. The cells were harvested and fixed with 25 mL/L glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for 2 h at 4 °C. For scanning electron microscopy (SEM) examination, the specimens were postfixed for 1 h in 2% OsO4, dehydrated by adding a series of graded ethanol solutions to the filtration system and then slowly dried over the course of 24 h by evaporation. The filter was removed from the filtration apparatus and mounted on an aluminum stub, after which the cells were gold sputter coated. Specimens were examined with a STEREOSCAN 260 SEM at 25 kV. For transmission electron microscopy (TEM) examination, cells corresponding to each population were collected in Haemoline (BioChem Pharma, Allentown, PA, USA), transferred to microcentrifuge tubes, pelleted, and fixed in 1% OsO4 (in distilled H2O). A total of 4×107 cells were sorted to collect 2×106 cells representative of each of the individual populations. After dehydration through a series of graded alcohol and propylene oxide solutions, the cells were infiltrated with Epon (epoxy resin) and polymerized. Ultra-thin sections were cut, recovered on Formvar-coated copper grids, stained with uranyl acetate and lead citrate, and then examined with a 100 CXII transmission electron microscope (Jeol, Tokyo, Japan) operated at 80 kV.
Cell proliferation assay The inhibition of [3H]TdR incorporation into DNA was examined using the pulse-labeling method. Proliferating HeLa cells were seeded onto 96-well plates and incubated for 16 h. HeLa cells were treated with different concentrations of spider venom (0, 10, 20,and 40 mg/L) for the next 24, 48, or 72 h. [3H]TdR (37 MBq/L) was added and HeLa cells were exposed to [3H]TdR for 16 h. The cells were then washed three times with PBS, lysed with 1 mol NaOH. After that the cells were harvested onto glass fiber filters with an automatic harvester. Filters were dried and radioactivity was quantified using liquid scintillation counter (Beckman LS6500, Fullerton, CA, USA). Results were expressed as the percent of specific lysis. Percent of specific lysis=100×[percent released from the target in the presence of effectors (experimental release)–percent released from the target in the presence of medium only (spontaneous release)]/(maximal release–spontaneous release).
Cell cytotoxicity assay Lactate dehydrogenase (LDH) release from cells was used as an index of cytotoxicity or necrosis. The quantity of LDH released by the cells into the medium was measured by the decrease in the absorbance at 340 nm for NADH disappearance within 5 min[13]. After incubation with various concentrations of spider venom (0, 10, 20, and 40 mg/L) for 24 h, the cell culture supernatant and medium (100 mL) were mixed with 900 mL PBS and 20 mg/L BSA. The percentage of LDH release (n=6) was equal to LDH activity in the medium divided by activity in both the cell culture supernatant and the medium×100%.
Cell cycle distribution using flow cytometry Bivariant flow cytometry was carried out on cells grown in the presence (control group) or absence of spider venom (10, 20, and 40 mg/L) after 24 h. Cells were collected and washed in cold PBS twice and resuspended in 100 mL of binding buffer (HEPES containing 2.5 mmol/L CaCl2). Fluorescein-labeled annexin V and PI were added to the cell suspension. Cells were then analyzed using flow cytometry (Becton Dickinson, Mountain View, CA, USA). The DNA content of the cells was determined by staining with PI. The cells were incubated in 100 mL of fixing solution (PBS containing 4% formaldehyde) for 15 min at 4 ºC, washed in PBS, resuspended in permeabilizing solution (PBS containing 0.1% saponin and 0.1% sodium azide) in the presence of 10 mL of PI, and incubated at 4 ºC for 15 min. The cells were then washed with PBS and immediately analyzed using flow cytometry.
Western blot analysis HeLa cells exposed to different concentrations of spider venom (10, 20, 40 mg/L and control) were incubated in six-well plates at a density of 2.5×105 cells/well for 48 h, followed by a further cultivation of 48 h with the culture medium replaced with 1 mL of serum-free RPMI-1640. The serum-free medium was totally collected. Equal amounts of proteins in each sample were resolved in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and the proteins were transferred onto nitrocellulose membranes. After being washed in 10 ml/L fat-free milk, the membranes were incubated with the appropriate dilution of rabbit polyclonal caspase-3 antibody and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody. The membranes were then incubated with a horseradish peroxidase-conjugated secondary antibody. Proteins were detected using an enhanced chemiluminescence (ECL) kit according to the manufacturer’s protocol (Amersham, Amersham, Buckinghamshire, UK).
In vivo reduction in tumor size in nude mice using spider venom We induced cervical tumors by injecting HeLa cells (200 µL) under the skin of nude mice with a concentration of 4×109 cells/L. Three weeks after tumor growth was established, treatment was initiated. Nude mice with tumors were randomly placed into three groups (10 mice/group) and treated with spider venom, 1.0, 2.0, 4.0 µg/g body weight of mice three times per week for 3 weeks by tail vein injections. The control group of 10 mice received distilled water only as a placebo. The mice were killed and tumor size was evaluated by caliper measurements, and tumor volume was calculated as length×width×depth.
Statistical analyses Significant differences between groups were tested using Mann-Whitney U tests with a significance level of 95%. Differences between groups were considered statistically significant at P<0.05. Analyses were carried our using the Prism software package (GraphPad, San Diego, CA, USA).
Results
Effects of spider venom on viability and apoptosis in HeLa cells Our study was carried out using HeLa cells grown in serum-free monolayer cultures. We observed using light microscopy that treatment with spider venom (10, 20, and 40 mg/L) initially produced clusters of packed cells (data not shown). After 24 h of treatment, the bulk of the cells appeared to be seriously damaged. To investigate the type of cell death induced by spider venom, the cells were stained with AO/EB, which allows the identification of viable, apoptotic and necrotic cells based on color and appearance. Staining with AO/EB of the samples treated with different concentrations of spider venom (Figure 1) showed different percentages of orange-stained cells. Viable cells were green with intact nuclei. Non-viable cells had bright orange chromatin. Apoptosis was demonstrated by the appearance of cell shrinkage with condensation and breaking up of the nuclei. Apoptotic cells were easily distinguished from necrotic cells because the latter appeared orange with a normal nuclear structure. This procedure was used to quantify the number of apoptotic cells after treatment with spider venom. A number of cells exhibited a flattened polygonal morpho-logy, whereas other cells showed morphological features of apoptosis, consisting of a decrease in cell volume, membrane broken, and breaking up of the nuclei. Moreover, the majority of these non-viable cells, together with a number of viable cells, showed broken nuclei and other signs of apoptosis.
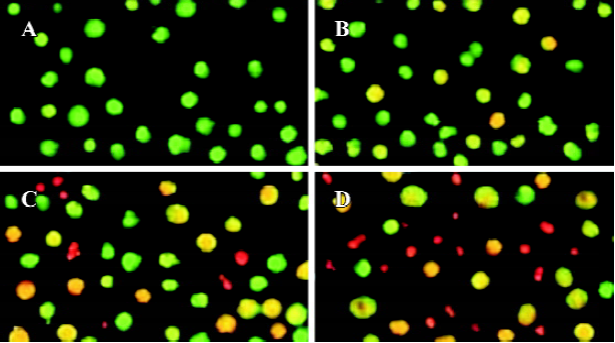
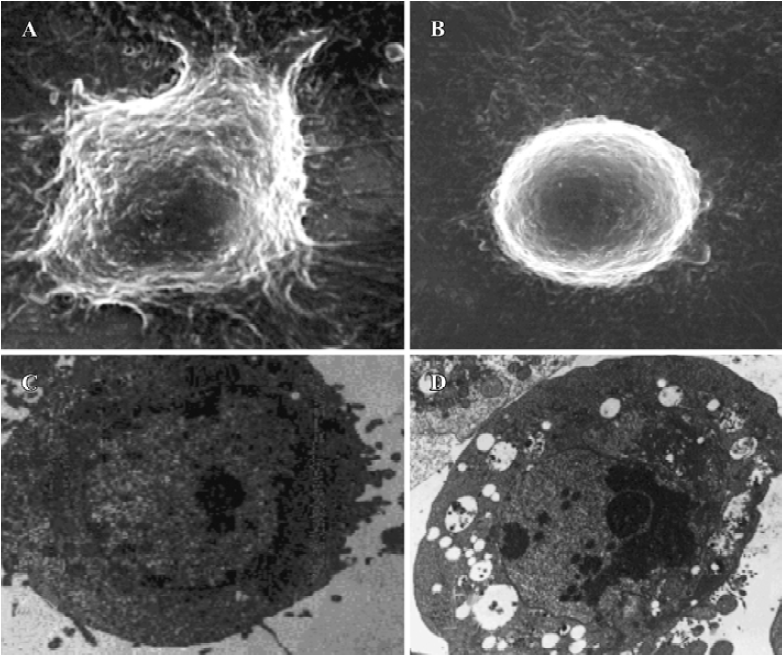
Morphological observations After exposure to spider venom for 24 h, SEM confirmed that HeLa cells treated with spider venom had morphological features indicative of apoptosis (Figure 2A,2B). Compared to cells treated with control supernatants, approximately 25% of cells exposed to spider venom (20 mg/L) for 24 h appeared to be markedly reduced in size. These cells were devoid of the villous projections found on the surfaces of control cells. In addition, many of the spider-venom-treated cells exhibited perturbations of their plasma membranes as evidenced by membrane smoothing. Ultrastructural characterization of cells representative of the individual populations was achieved through cell sorting and subsequent evaluation using TEM. Cells treated with spider venom possessed well-defined plasma membranes and contained intact organelles with no evidence of nuclear condensation (Figure 2C). In contrast, the majority of cells in the venom-treated group exhibited morphological characteristics typical of cells in the early stages of apoptosis. These characteristics included a decrease in cell size, intact cell membranes, and flocculation of the nucleus (Figure 2D). None of these characteristics were present in untreated cells.
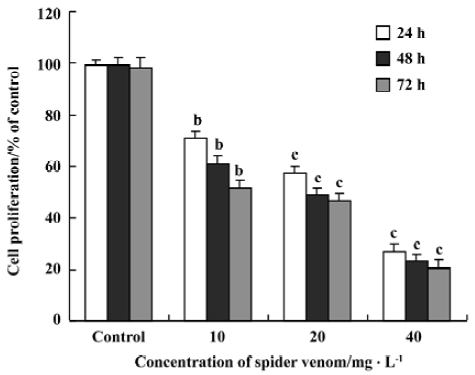
Effect of spider venom on [3H]TdR incorporation in HeLa cells Inhibition of DNA synthesis was examined in the presence of spider venom (Figure 3). Proliferating HeLa cells were exposed to different concentrations of spider venom for 24, 48, or 72 h. The cells were then treated with [3H]TdR for 12 h. Synthesis of DNA in these cells was inhibited, with IC50 ranging from 20 to 30 mg/L after 24 h, 48 h, 72 h. The degree of growth inhibition of HeLa cells in the presence of spider venom was dose-dependent. Spider venom significantly decreased cell proliferation, but the degree differed with the variant concentrations (P<0.01). The spider-venom-induced inhibition of cell proliferation was also time dependent. Spider venom significantly decreased cell proliferation, and again the decrease differed with time (P<0.01).
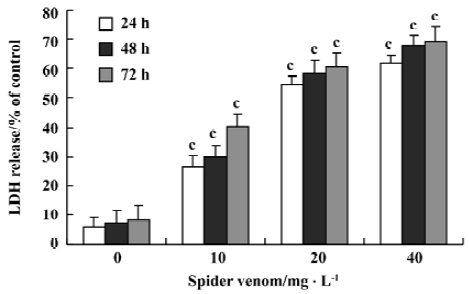
Cell cytotoxicity of spider venom on HeLa cells Cell cytotoxicity was directly measured by LDH release (Figure 4). After 24 h incubation, spider venom (0, 10, 20, and 40 mg/L) significantly increased cell cytotoxicity (P<0.05) compared with the control group (5.95%). Increased cell cytotoxicity occurred with increased concentrations of spider venom. The activity is to be time- and dose-dependent.
Effect of spider venom on apoptosis, necrosis ratio, and cell cycle Cells were exposed to different concentrations of spider venom (10, 20, and 40 mg/L). Cells in the early phase of apoptosis were labeled using single annexin V fluorescence. The ratio of apoptosis and necrosis in the cells increased with increasing venom concentrations. The early stages of apoptosis and necrosis started 5 h after treatment of HeLa cells with 40 mg/L spider venom (Table 1).
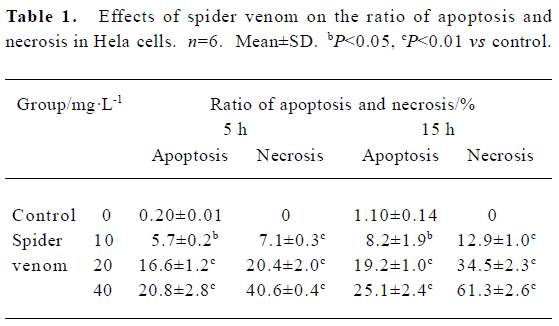
Full table
Cell cycle distribution, cell proliferation and apoptotic damage of DNA were determined using a flow cytometer. Results showed the accumulation of cells in S and G2/M, and a corresponding reduction in the percentages of cells in G0/G1 (Table 2).
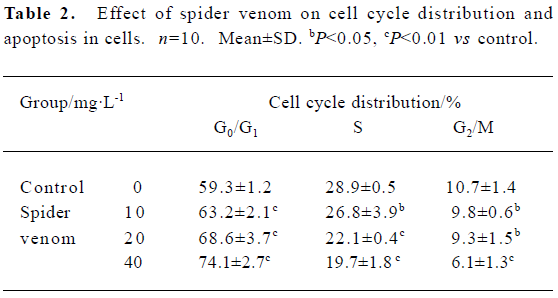
Full table
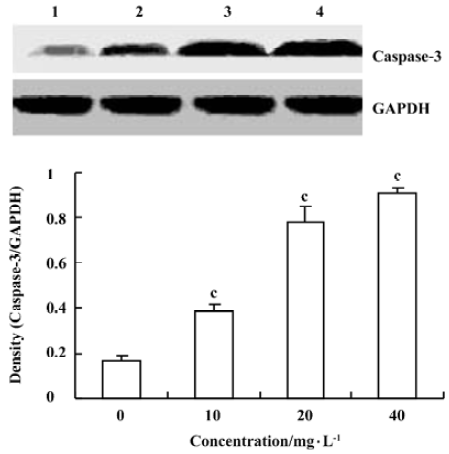
Effects of spider venom on the activitiy of caspase-3 To elucidate the pathway leading to apoptosis, we examined the activation of caspase-3, which was reported to initiate apoptosis after various stimuli. HeLa cells treated with spider venom (10, 20, and 40 mg/L) for 48 h were analyzed for enzymatic activity using a Western blot analysis. Caspase-3 activity changed at 48 h in HeLa cells (Figure 5). The activity was enhanced significantly (1.7-, 2.2-, and 4.3-fold, respectively, versus controls, P<0.01). Western blotting analysis, revealed that the exposure of HeLa cells to spider venom enhanced the activity of caspase-3, which became four-fold higher than that in the control cells (Figure 5).
In vivo reduction in tumor size in nude mice using spider venom We have investigated the effect of spider venom in nude mice with tumors. Mice were killed 21 d after treatment and tumor size was determined. In the groups injected with various concentrations of spider venom by the tail vein, the size of the tumor inside the skin was significantly smaller (P<0.01) than in untreated mice (Table 3).
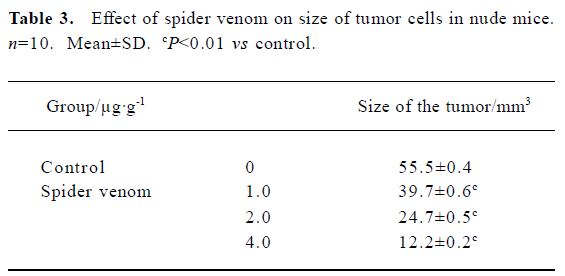
Full table
Discussion
A number of possible biological and biochemical effects of spider venom were investigated in the present study.
Cells excluding vital dyes were considered to be viable. However, our current results suggest that many of these cells are actually undergoing apoptotic cell death despite having relatively intact cell membranes. Spider venom at doses of 40, 20, and 10 mg/L significantly decreased cell proliferation in HeLa cells, in a dose-dependent and time-dependent manner.
Aside from the inhibitory effect of spider venom on cell proliferation, spider venom at the same doses (10, 20, and 40 mg/L) significantly increased cytotoxicity determined by LDH release in the cells. The cytotoxic effect of spider venom reached a plateau at doses over 160 mg/L. The cytotoxic effect of spider venom could be attributed to necrosis[24]. The total number of cells present will be directly proportional to the background-subtracted fluorescence values, which represent LDH activity. It is relatively easy to distinguish between complete cell lysis occurring at time zero versus 50% cell lysis at 24 h, 48 h, and 72 h and this result was observed in the “lysed”data (Figure 4).
Meanwhile, we directly measured apoptosis using flow cytometry. Apoptosis in HeLa cells was increased in the spider venom-treated groups. It is possible that apoptosis contributes to the cytotoxic effect of spider venom. Changes in cells associated with the early phases of apoptosis include a loss of cell membrane phospholipid symmetry. The effect of spider venom on HeLa cells was analyzed using fluorescein-labelled annexin V-stained and PI-stained cells (Table 1). Thus, our findings revealed that spider venom resulted in direct necrosis and indirect apoptosis (Table 1) to kill HeLa cells.
To examine whether growth inhibition of HeLa cells by spider venom resulted from cell cycle arrest, cell cycles treated with spider venom were analyzed using flow cytometry. The inhibitory effect of spider venom on growth of the cells may result from G0/G1 cell cycle arrest.
Caspases are common and critical components of the cell death pathway. Among these caspases, caspase-3-like proteases mediate the initiation and/or execution stages of programmed cell death[25,26]. The active enzyme takes part in the execution phase of apoptosis and it has been demonstrated that a high level of activity of effector caspase-3 in tumor cells plays a decisive role in their commitment to apoptosis[27–30]. Caspase-3 is a key executioner of apoptosis, whose activation is mediated by the initiator caspases, such as caspase-9[31]. Spider-venom-induced apoptosis was preceded by the activation of caspase-3, which was clearly observed after 48 h of treatment by direct estimation of its activity and by Western blotting analysis, which showed the conversion of procaspase-3 to the active form of the enzyme. This result is the same as norcantharidin-induced HeLa cell apoptosis resulting from an increase in caspase-3 activity[32].
All these events preceded the appearance of the morphological signs of apoptosis, which were observed in a large percentage of cells only after 48 h of treatment (Figure 5). The results from the present study suggested that the cytotoxic effect of spider venom on the cells was related to the induction of apoptosis.
Importantly, the anti-tumor in vivo action of spider venom resulted in either complete or significant regression of the human cervical tumors established in nude mice. Our in vivo results demonstrate that spider venom confers a systemic effect of tumor regression in a xenograft in a dose-dependent manner.
In conclusion, we demonstrated that spider venom caused an inhibition of cell growth in HeLa cells as a result of cycle arrest and apoptosis. Moreover, a large part of our study essentially focused on the mitochondrial pathway and we determined that one action of spider venom was caspase-3 dependent. These new findings suggest that spider-venom-induced effects may have novel applications for the treatment of cancer.
Acknowledgements
We are indebted to Jun-xin LI (Research Centre, the Fourth Hospital of Hebei Medical University) for equipment and technical assistance. We are also grateful to Ji-biao LI for technical assistance with the SEM; Jian ZHANG for molecular technical assistance and sharing his expertise (Hebei Normal University, Shijiazhuang, China). I thank Dr James P KEHRER (University of Texas at Austin,TX, USA) for helping us improve the manuscript.
References
- Schanbacher FL, Lee CK, Hall JE, Wilson IB, Howell DE, Odell GV. Composition and properties of tarantula Dugesiella hentzi (Girard) venom. Toxicon 1973;11:21-4.
- Jackson H, Parks TN. Spider toxins: recent applications in neurobiology. Annu Rev Neurosci 1989;12:405-14.
- Ori M, Ikeda H. Spider venoms and spider toxins. J Toxicol 1998;17:405-26.
- Rash LD, Hodgson WC. Pharmacology and biochemistry of spider venoms. Toxicon 2002;40:225-54.
- Abu-Sinna G, Esmat AY, Al-Zahaby AA, Soliman NA, Ibrahim TM. Fractionation and characterization of Cerastes cerastes snake venom and the antitumor action of its lethal and non-lethal fractions. Toxicon 2003;42:207-15.
- Swenson S, Costa F, Minea R, Sherwin RP, Ernst W, Fujii G, et al. Intravenous liposomal delivery of the snake venom disintegrin contortrostatin limits breast cancer progression. Mol Cancer Ther 2004;3:499-511.
- Correa MC Jr, Maria DA, Moura-da-Silva AM, Pizzocaro KF, Ruiz IR. Inhibition of melanoma cell tumorigenicity by the snake venom toxin jararhagin. Toxicon 2002;40:739-48.
- de Carvalho DD, Schmitmeier S, Novello JC, Markland FS. Effect of BJcuL (a lectin from the venom of the snake Bothrops jararacussu) on adhesion and growth of tumor and endothelial cells. Toxicon 2001;39:1471-6.
- Schmitmeier S, Markland FS, Chen TC. Anti-invasive effect of contortrostatin, a snake venom disintegrin, and TNF-alpha on malignant glioma cells. Anticancer Res 2000;20:4227-33.
- Orsolic N, Sver L, Verstovsek S, Terzic S, Basic I. Inhibition of mammary carcinoma cell proliferation in vitro and tumor growth in vivo by bee venom. Toxicon 2003;41:861-70.
- Shaposhnikova VV, Kublik LN, Narimanov AA, Levitman MKh, Orlova OE, Kudriavtsev AA, et al. Growth inhibition and induction of tumor cell death by phospholipase A2 and lipoxygenase inhibitors. Izv Akad Nauk Ser Biol 2001.249-52.
- Liu X, Chen D, Xie L, Zhang R. Effect of honey bee venom on proliferation of K1735M2 mouse melanoma cells in-vitro and growth of murine B16 melanomas in-vivo. J Pharm Pharmacol 2002;54:1083-9.
- Liu YF, Ma RL, Wang SL, Duan ZY, Zhang JH, Wu LJ, et al. Expression of an antitumor-analgesic peptide from the venom of Chinese scorpion Buthus martensii karsch in Escherichia coli. Protein Expr Purif 2003;27:253-8.
- Szeto TH, Birinyi-Strachan LC, Smith R, Connor M, Christie MJ, King GF, et al. Isolation and pharmacological characterisation of delta-atracotoxin-Hv1b, a vertebrate- selective sodium channel toxin. FEBS Lett 2000;470:293-9.
- Stotz SC. Block of voltage-dependent calcium channel by the green mamba toxin calcicludine. J Membrane Biol 2000;174:157-65.
- Marvin L, De E, Cosette P, Gagnon J, Molle G, Lange C. Isolation, amino acid sequence and functional assays of SGTx1. The first toxin purified from the venom of the spider Scodra griseipes. Eur J Biochem 1999;265:572-9.
- Corzo G, Gilles N, Satake H, Villegas E, Dai L, Nakajima T, et al. Distinct primary structures of the major peptide toxins from the venom of the spider Macrothele gigas that bind to sites 3 and 4 in the sodium channel. FEBS Lett 2003;547:43-50.
- Satake H, Villegas E, Oshiro N, Terada K, Shinada T, Corzo G. Rapid and efficient identification of cysteine-rich peptides by random screening of a venom gland cDNA library from the hexathelid spider Macrothele gigas. Toxicon 2004;44:149-56.
- Zhu MS, Li TH, Song DX. A species of genus macrothele (araneae: hexathekidae) from China. J Hebei Univ 2000;4:358-61. (Natural Science Edition).
- Zheng XZ, Liang SP. Purification and preliminary toxin research of raventoxin-II, a neurotoxic peptide from the venom of the spider Macrothele raveni. Life Sci Res 2001;3:217-20.
- Zeng XZ, Xiao QB, Liang SP. Purification and characterization of raventoxin-I and raventoxin-III, two neurotoxic peptides from the venom of the spider Macrothele raveni. Toxicon 2003;41:651-6.
- Friedel T, Nentwig W. Immobilizing and lethal effects of spider venoms on the cockroach and the common mealbeetle. Toxicon 1989;27:305-16.
- Giuliano M, Lauricella M, Vassallo E, Carabillò M, Vento R, Tesoriere G. Induction of apoptosis in human retinoblastoma cells by topoisomerase inhibitors. Invest Ophthalmol Vis Sci 1998;39:1300-11.
- Zhang Y, Wu LJ, Tashiro S, Onodera S, Ikejima T. Evodiamine induces tumor cell death through different pathways: apoptosis and necrosis. Acta Pharmacol Sin 2004;25:83-9.
- Qi H, Chen HZ, Jin ZJ. Caspase 3 gene expression and [Ca2+]i homeostasis underlying desipramine-induced C6 glioma cell apoptosis. Acta Pharmacol Sin 2002;23:803-7.
- Lan H, Lu YY. Allitridi induces apoptosis by affecting Bcl-2 expression and caspase-3 activity in human gastric cancer cells. Acta Pharmacol Sin 2004;25:219-25.
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, et al. Human ICE/CED-3 protease nomenclature. Cell 1996;87:171.
- Chae HJ, Park KM, Lee GY, Jeong GS, Park HR, Kim HM, et al. Je-chun-jun induced apoptosis of human cervical carcinoma HeLa cells. Acta Pharmacol Sin 2004;25:1372-9.
- Liu DC, Zang CB, Liu HY, Possinger K, Fan SG, Elstner E. A novel PPAR alpha/gamma dual agonist inhibits cell growth and induces apoptosis in human glioblastoma T98G cells. Acta Pharmacol Sin 2004;25:1312-19.
- Hou R, Zhou QL, Wang BX, Tashiro S, Onodera S, Ikejima T. Diosgenin induces apoptosis in HeLa cells via activation of caspase pathway. Acta Pharmacol Sin 2004;25:1077-82.
- Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell 1997;91:443-6.
- An WW, Gong XF, Wang MW, Tashiro S, Onodera S, Ikejima T. Norcantharidin induces apoptosis in HeLa cells through caspase, MAPK, and mitochondrial pathways. Acta Pharmacol Sin 2004;25:1502-8.