
Protective effects of cariporide on endothelial dysfunction induced by high glucose1
Introduction
The relation between diabetes and premature vascular disease has been well established[1]. One of the defects involves endothelial dysfunction characterized by impaired endothelium-dependent relaxation responses. Many metabolic disturbances of diabetes, hyperglycemia have been suggested to be the main cause of endothelial dysfunction. High glucose in vitro or in vivo has been reported to inhibit acetylcholine (ACh)-induced endothelium-dependent relaxation[2] responses, to impair the biological synthesis pathway of nitric oxide (NO)[3], and to generate reactive oxygen species[4].
It has been demonstrated that hyperactivity of sodium-hydrogen exchange subtype 1 (NHE-1) has been implicated in the vascular injury in diabetes mellitus[5]. Ganz et al[6] found that the activity and expression of NHE-1 significantly increased in mesangial cells after exposure to high glucose. Our previous experiments have demonstrated that the benzoylguanidine compound cariporide (4-isopropyl-3-methylsulfonyl-benzoylguanidine methanesulfonate), which is a selective NHE-1 inhibitor, protected against injuries of endothelial functions induced by high lipid diet in rabbits[7]. However, it is not known whether NHE-1 inhibitor protects against endothelial function affected by high glucose. The aim of this study is to explore the effect of cariporide against endothelial dysfunction of isolated rat aortic rings induced by high glucose and to investigate its mechanisms.
Materials and methods
Drugs and chemicals Cariporide was obtained from Hoechst Company (Frankfurt, Germany). SNP, ACh, and phenylephrine (Phe) were purchased from Sigma Chemical Co (Saint Louis, Mo, USA). The kits for measurement of nitrite/nitrate (NO), MDA, and SOD activity were purchased from Nanjing Jiancheng Bioengineering Institute (Jiangsu, China).
Animal Male Sprague-Dawley rats (N
Preparation of rat thoracic aorta rings The rats were killed by exsanguinations after an intraperitoneal anesthesia with pentobarbital sodium 30 mg/kg and intravenous anticoagulation with heparin sodium 150 U/kg. The descending thoracic aorta was rapidly dissected from the rats and immersed in Krebs’ solution, composed of (mmol/L): NaCl, 118.3; KCl, 4.7; MgSO4·7H2O, 1.2; KH2PO4, 1.2; CaCl2, 2.5; NaHCO3, 24.0; glucose, 11; and Na2-EDTA, 0.026; and bubbled with 95% O2+5% CO2 (pH 7.4). After the perivascular tissue was carefully removed, the aortic rings (which were approximately 4 mm in length), were prepared.
Bioassay of vasoreactivity[8] For isometric force record-ing, the aortic rings were mounted between two stainless steel hooks and suspended in a 10 mL organ bath containing above Krebs’ solution at 37 ºC bubbled with 95% O2+5% CO2 gas mixture (pH 7.4). An initial load of 2.0 g was applied, and the tension of the aortic ring was monitored by a force transducer and recorded on a polygraph (Model YL-1, Chengdu Instruments, China). After a 90-min equilibration period, the ring was precontracted by Phe 1 µmol/L. When the development tension attained its peak value, the ring was relaxed by ACh or SNP, respectively. Accumulative concentration-response curves to ACh (0.003, 0.01, 0.03, 0.1, 0.3, 1, and 3 µmol/L) or SNP (0.001-1 µmol/L) were recorded.
Determination of MDA concentration After a 6-h incubation of aortic segments, the aortic segments were blotted dry and weighed, then made into 5% tissue homogenate in ice-cold 0.9% NaCl solution. A supernatant was obtained from tissue homogenate by centrifugalization (1000×g, 4 ºC, 10 min). The MDA concentration (thiobarbituric acid reactive substances, TBARS) in the supernatant was measured. Briefly, 1.0 mL of 20% trichloroacetic acid and 1.0 mL of 1% TBARS reagent were added to 100 µL supernatant, then mixed and incubated at 100 ºC for 80 min. After cooling on ice, samples were centrifuged at 1000×g for 20 min and the absorbance of the supernatant was read at 532 nm. TBARS results were expressed as MDA equivalents using tetraethoxy-propane as standard.
Assay of SOD activity in aortic rings The supernatant of tissue homogenate of the aortic rings were obtained as described earlier. A competitive inhibition assay was performed by using xanthine/xanthine oxidase reaction-generated superoxide radicals to reduce nitro blue tetrazolium (NBT) quantitatively to blue formazan. Conversion of superoxide radicals to hydrogen peroxide by superoxide dismutase inhibited dye formation and served as a measure of superoxide dismutase activity. Briefly, the supernatant of 0.5 mL with xanthine 50 µmol/L and xanthine oxidase 2.5 µmol/L in potassium phosphate buffer 50 mmol/L (pH 7.8, 37 ºC) were incubated for 40 min and NBT was added. Blue formazan was then monitored spectrophotometrically at 550 nm. The amount of protein that inhibited NBT reduction to 50% maximum was defined as 1 nitrite unit (NU) of SOD activity.
Assay of NO concentration of incubation medium The incubation medium of the aortic artery was centrifugated (1000×g, 15 min, 4 ºC) and the supernatant was used for NO measurement. NO was assayed by the Griess method. Because NO is a compound with a short half life and is rapidly converted to the stable end products nitrate (NO3–) and nitrite (NO2–), the principle of the assay is the conversion of nitrate into nitrite by cadmium and followed by color development with Griess reagent (sulfanilamide and N-naphthyl ethylenediamine) in acidic medium. The total nitrite was measured by Griess reaction. The absorbance was determined at 540 nm with a spectrophotometer.
Protocol of experiment The first series of experiments were designed in order to evaluate the protective effects of cariporide against ACh-induced endothelium-dependent and SNP-induced endothelium-independent relaxing response of isolating rat aortic rings affected by high glucose. The experiment was divided into 7 groups with 8 aortic rings from 8 rats in each group. First, a normal control bioassay of vasoreactivity was formed in normal Krebs’ solution. The rings, of which a percentage of relaxation induced by ACh 3 µmol/L to the contraction elicited by Phe 1 µmol/L is more than 80%, were considered as intact endothelium and used in the study. The aortic rings of each group were then continually incubated for 6 h in the following medium: (1) control group: glucose 11 mmol/L in Krebs’ solution[9]; (2) high glucose group: glucose 44 mmol/L in Krebs’ solution[9]; (3)–(5) cariporide-treated groups: cariporide 0.01, 0.1, and 1 µmol/L in Krebs’ solution with glucose 44 mmol/L, respectively; (6) cariporide alone group: cariporide 1 µmol/L in Krebs’ solution; (7) mannitol group: mannitol 44 mmol/L in Krebs’ solution. The incubation mediums were changed every 30 min and cariporide was present throughout the incubation. After a 6-h incubation of aortic rings, the perfusion solution was changed to Krebs’ solution, and bioassay of vasore-activity was performed.
The second series of experiments were designed to assay the effects of high glucose on SOD activity, MDA concentration, NO and the effects of cariporide on the biochemical parameters in rat aortic rings. The experiment was divided into 5 groups with 8 aortic segments of 2 cm from 8 rats in each group; control group, high glucose group, mannitol group, cariporide-treated group, and cariporide-alone group. The components of incubation medium were the same as described earlier except that cariporide only had a dose of 1 µmol/L. The incubations media were changed every 30 min. Cariporide was added throughout incubation. After a 6-h incubation, the segments were transfered to 1 mL normal Kreb’s solution which contained ACh 1 µmol/L for 30 min. The aortic segments and the medium were then collected and frozen at -70 ºC until analyzed.
Data analysis The ACh (3 µmol/L) or SNP (1 µmol/L) -induced maximal relaxation (Emax) in aortic rings was calculated as a percentage of the contraction to Phe (1 µmol/L). The half maximum effective concentration (EC50) was defined as a concentration of the ACh that induced 50% of maximum relaxation response to contraction elicited by Phe (1 µmol/L) and calculated from the concentration-response curve generated by linear regression analysis. All data were expressed as mean±SD. Statistical comparisons were made using one-way ANOVA followed by Newman–Keuls test. P<0.05 was statistically significant.
Results
Effects of high glucose on EDR and endothelium-independent relaxation in aortic rings There were no significant differences in ACh-induced relaxation responses of rat isolated aortic rings before the 6-h incubation among various groups (data not shown). After a 6-h incubation of aortic rings in control glucose (11 mmol/L) buffer, ACh (0.003-3 µmol/L) still evoked a normal concentration-dependent relaxation (Figure 1), the Emax of aortic rings reached 88.4%±12.3%, and the EC50 value was 94.5±10.8 nmol/L. After the 6-h incubation of the aortic rings and exposure to high glucose (44 mmol/L), the Emax fell to 43.7%±16.1% and the EC50 value increased to 154.8±22.9 nmol/L (P<0.01 vs control group, n=8). However, no significant changes of Emax and EC50 were shown in the rings incubated in the cariporide (1 µmol/L)-alone group or mannitol (44 mmol/L) group, compared with the control group (Figure 1).
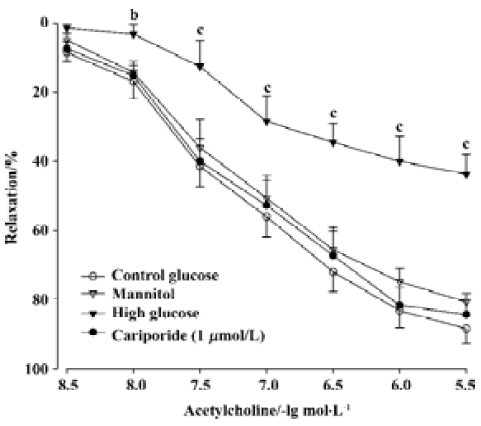
The endothelium-independent relaxation induced by SNP (0.001-1 µmol/L) was not significantly different between the different treated groups (data not shown).
Effects of cariporide on the impairment of EDR induced by high glucose Treatment with cariporide in different concentration (0.01, 0.1, and 1 µmol/L, n=8, respectively) significantly prevented inhibition of EDR induced by high glucose (Figure 2). The Emax were 61.7%±10.5%, 76.0%±10.5%, and 83.4%±10.1%, and the EC50 value was 131.5±15.9 nmol/L, 117.1±13.7 nmol/L, and 109.6±10.5 nmol/L, respectively. There was a significant difference (P<0.05 or P<0.01), compared with those in the high glucose group (Figure 2).
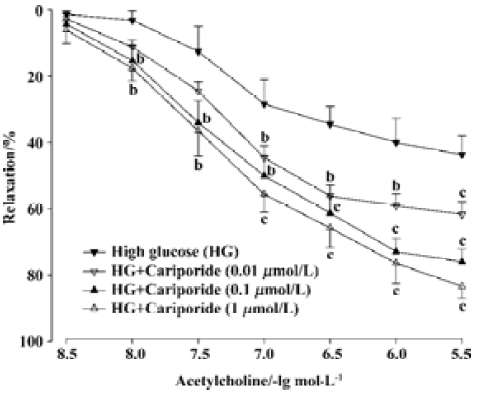
Effects of cariporide on biochemical index in aortic segments A 6-h incubation of isolated aortic segments in high glucose resulted in an elevation of MDA content, decrease of SOD activity in aortic tissue, and reduction of NO releasing from aortic segments. Treatment with cariporide (1 µmol/L) in the high glucose (44 mmol/L) group significantly prevented the increase of MDA content, and protected the activity of SOD and release of NO in aortic segments (Table 1). Mannitol (44 mmol/L) or cariporide (1 µmol/L) alone had no effects on MDA, SOD, and NO, compared with the control group (Table 1).
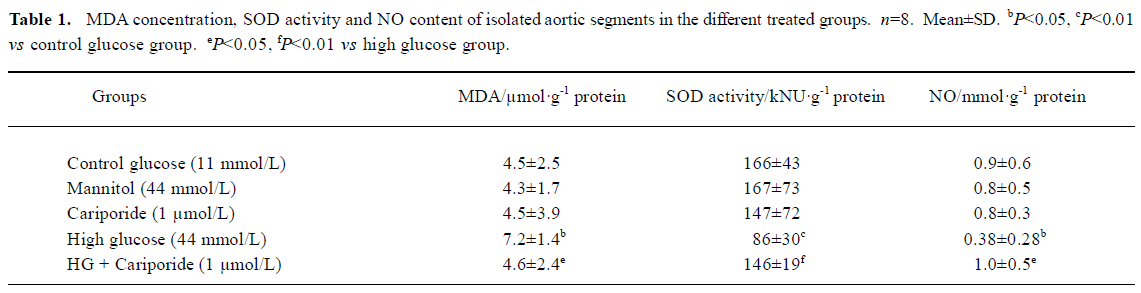
Full table
Discussion
Diabetes mellitus is characterized by chronic hyperglycemia and associated with significant morbidity as a result of long-term complications, including diabetic nephropathy, atherosclerosis, and hypertension. High glucose had a lot of toxicity effects in endothelial cells, such as impairment of endothelial dependent relaxation[10], decrease of NO release, generation of free radicals, and increase in apoptosis[11]. The endothelial dysfunction has been thought to be the major cause of vascular disease due to hyperglycemia condition. It was reported that hyperglycemia resulted in an increase of NHE-1 activity in diabetes[12,13]. In addition, high glucose may induce the activation of NHE-1 and increase NHE-1 mRNA expression in vitro smooth muscle cells or myocytes [14,15]. However, there are no reports as to whether NHE-1 inhibitor is able to protect against the impairment of endothelial functions caused by high glucose. The present study explores the effects of cariporide against high glucose-induced endothelial dysfunction of rat isolated aorta and discusses its mechanisms.
In the present study, we employed a mimic pronounced hyperglycemia model in which the rat isolated aortic rings were exposed to high glucose for 6 h. High glucose significantly inhibited EDR and the release of NO in rat isolated aortic rings, but did not affect vasodilatation induced by SNP, a NO donor. It has been demonstrated that endothelium dysfunction induced by high glucose related to the release of NO from endothelial cells. The same concentration of mannitol had no effect on EDR of aortic rings, which showed that the damage of EDR of aortic rings induced by high glucose was not due to a hyperosmotic effect. These results in the present study were consistent with a previous study[2]. We also found that cariporide in dose-dependent manners prevented the inhibition of EDR induced by high glucose in rat isolated thoracic aorta (Figure 1), and simultaneously maintained SOD activity and NO release and decreased MDA concentration caused by high glucose in rat aortic segments (Table 1). It was reported that cariporide significantly inhibited the injuries of mitochondrial and pulmonary endothelial cells, and protected cardiac ischaemia and reperfusion in vitro by reducing intracellular pH and inhibiting NHE-1 activation, which simultaneously attenuated oxidant production[16-18]. The Na+/H+ exchanger inhibitor, amiloride, significantly reduced oxidant production of hepatic stellate cells including intracellular hydroperoxides and MDA induced by oxidant[19]. Accordingly, it is believed that the protective effects of cariporide against endothelial dysfunctions induced by high glucose may partly be due to anti-oxidation and protective activity of anti-oxidative enzymes.
In the resting state, NHE-1 is relatively quiescent. When intracellular pH falls, the NHE-1 is activated, and the rate of H+ efflux mediated by NHE-1 increases and causes intracellular alkalinization and Na+ overload, which leads to intracellular Ca2+ overload through a mechanism of reverse Na+-Ca2+ exchange. It is presumed that during high glucose condition, there is a metabolic mismatch between glycolysis and glucose oxidation that results in the accumulation of hydrogen ions, which, in turn, activates NHE-1, leading to intracellular alkalinization and Ca2+ overload. Therefore, we hypothesized that, in the present study, the mechanisms of cariporide against high glucose-induced endothelial dysfunction of rat isolated aortas might be also related to decreasing intracellular alkalinization and Ca2+ overload. Although intracellular pH and intracellular Ca2+ concentration were not assayed in the study, it has been reported that cariporide could protect from releasing NO in vascular endothelium through inhibiting activity of NHE-1, decreasing intracellular alkalinization and Ca2+ overload[17,20].
In conclusion, this study demonstrates that cariporide significantly prevents endothelial dysfunction, decreases NO release, elevates MDA concentration and reduces SOD activity induced by high glucose in rat isolated thoracic aorta. The mechanisms of protective effects of cariporide may be related to the inhibition of NHE-1 and the decrease of oxidative stress injury. These results provide a potential new target for intervention in the prevention of diabetic complica-tions.
References
- Najemnik C, Sinzinger H, Kritz H. Endothelial dysfunction, atherosclerosis and diabetes. Acta Med Austriaca 1999;26:148-53.
- Guo X, Liu WL, Chen LW, Guo ZG. High glucose impairs endothelium-dependent relaxation in rabbit aorta. Acta Pharmacol Sin 2000;21:169-73.
- Pieper GM. Enhanced, unaltered and impaired nitric oxide-mediated endothelium-dependent relaxation in experimental diabetes mellitus: importance of disease duration. Diabetologia 1999;42:204-13.
- Friesen NT, Buchau AS, Schott-Ohly P, Lgssiar A, Gleichmann H. Generation of hydrogen peroxide and failure of antioxidative responses in pancreatic islets of male C57BL/6 mice are associated with diabetes induced by multiple low doses of streptozotocin. Diabetologia 2004;47:676-85.
- Matteucci E. Giampietro. Sodium/hydrogen exchange activity in type 1 diabetes mellitus: the never-ending story. Diabetes Nutr Metab 2001;14:225-33.
- Ganz MB, Hawkins K, Reilly RF. High glucose induces the activity and expression of Na+/H+ exchange in glomerular mesangial cells. Am J Physiol Renal Physiol 2000;278:F91-6.
- Tu JH, Liu LY, Zhang XH. Protective effect of Na+/H+ exchanger inhibitor cariporide on the injury of vascular endothelial function induced by hypercholesterolemia. Bull Hu-nan Med Univ 2002;27:13-6.
- Ma FX, Liu LY, Xiong XM. Protective effects of lovastatin on vascular endothelium injured by low density lipoprotein. Acta Pharmacol Sin 2003;24:1027-32.
- Tesfamariam B, Brown ML, Cohen RA. Elevated glucose impaired endothelium-dependent relaxation by activating protein kinase C. J Clin Invest 1991;87:1643-8.
- Xu B, Rao MR. High concentration of glucose inhibits endothelium-dependent vasorelaxation of rabbit aortic artery. Acta Pharmacol Sin 2001;22:861-4.
- Guo X, Liu WL, Chen LW, Guo ZG. High glucose enhances H2O2-induced apoptosis in bovine aortic endothelial cells. Acta Pharmacol Sin 2000;21:41-5.
- Jandeleit-Dahm K, Hannan KM, Farrelly CA, Allen TJ, Rumble JR, Gilbert RE, et al. Diabetes-induced vascular hypertrophy is accompanied by activation of Na+/H+ exchange and prevented by Na+/H+ exchange inhibition. Circ Res 2000;87:1133-40.
- Telejko B, Tomasiak M, Stelmach H, Kinalska I. Platelet sodium-proton exchanger and phospholipid-dependent procoagulant activity in patients with type 2 diabetes. Metabolism 2003;52:102-6.
- Williams B, Howard RL. Glucose-induced changes in Na+/H+ antiport activity and gene expression in cultured vascular smooth muscle cells. Role of protein kinase C. J Clin Invest 1994;93:2623-31.
- Siczkowski M, Ng LL. Glucose-induced changes in activity and phosphorylation of the Na+/H+ exchanger, NHE-1, in vascular myocytes from Wistar-Kyoto and spontaneously hypertensive rats. Metabolism 1996;45:114-9.
- Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, et al. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res 1995;29:260-8.
- Teshima Y, Akao M, Jones SP, Marban E. Cariporide (HOE642), a selective Na+/H+ exchange inhibitor, inhibits the mitochondrial death pathway. Circulation 2003;108:2275-81.
- Cutaia M, Kroczynski J, Tollefson K. pH-dependent oxidant production following inhibition of the mitochondrial electron transport chain in pulmonary endothelial cells. Endothelium 2002;9:109-21.
- Svegliati-Baroni G, Di Sario A, Casini A, Ferretti G, D’Ambrosio L, Ridolfi F, Bolognini L, et al. The Na+/H+ exchanger modulates the fibrogenic effect of oxidative stress in rat hepatic stellate cells. J Hepatol 1999;30:868-75.
- Stromer H, de Groot MC, Horn M, Faul C, Leupold A, Morgan JP, et al. Na+/H+ exchange inhibition with HOE642 improves postischemic recovery due to attenuation of Ca2+ overload and prolonged acidosis on reperfusion. Circulation 2000;101:2749-55.