
Inducible nitric oxide synthase contributes to intermittent hypoxia against ischemia/reperfusion injury1
Introduction
Intermittent hypoxia, or periodic exposure to hypoxia interrupted by a return to normoxia or less hypoxic conditions, is encountered more frequently in life than sustained hypoxia[1,2]. Many studies have shown that intermittent hypoxia adaptation might have cardioprotective effects similar to those observed in ischemic preconditioning (IPC)[3–6]. To date, several potential factors have been proposed to be involved in the protective mechanisms afforded by IH[4–10]; however, the precise mechanisms in which IH increases resistance to myocardial ischemia remain far from clear.
Beall et al reported that exhalation of nitric oxide (NO) by chronically hypoxic populations of Tibetans and Bolivian Aymara is unexpectedly increased compared with low-altitude populations[11]. The similar response of these two geographically separate high-altitude populations underlines the importance of NO for life under hypoxic stress. Beall et al speculated that one possible adaptation to maintaining high-output NO synthesis under hypoxia included increased expression of the synthase enzymes themselves. Nitric oxide synthase (NOS) are a family of three isozymes responsible for the production of NO: the constitutive endothelial (eNOS) and neuronal (nNOS) isozymes and the inducible isozyme. It is known that iNOS is expressed in a wide variety of cell types, including cardiac myocytes. iNOS is usually expressed in response to various physiological and pathophysiological stimuli, such as intense exercise and hypoxia. Recent studies have shown that NO plays an important role in protecting myocardium from I/R injury[12–14]. In addition, studies have indicated that the cardioprotective effects of late preconditioning observed after 24 h resulted from the upregulation of NOS and, more specifically, of iNOS[15,16]. Neckar et al[17] reported that the cardioprotective effects of chronic hypoxia and IPC were not additive, suggesting that the mechanisms of protection conferred by chronic hypoxia and preconditioning may share some common signaling pathways. At present, little is known about the role of iNOS-derived NO in the cardioprotection of IH and there is no evidence about the changes in iNOS mRNA and protein in IH rat hearts subjected to ischemia and reperfusion. The aim of the present study was to evaluate: (i) the effect of aminoguanidine (AG) on the post-ischemic recovery of left ventricular function in IH rat hearts, thereby determining the role of iNOS-derived NO in the cardioprotection afforded by IH; and (ii) the effect of IH on iNOS mRNA and protein expression in rat hearts during baseline perfusion, ischemia and reperfusion period.
Materials and methods
Animal preparation Adult male Sprague-Dawley rats (Clean grade, Shanghai Experimental Animal Center, Chinese Academy of Sciences, Shanghai, China), initially weighing 100–130 g and finally weighing 280–330 g, were exposed to intermittent high-altitude hypoxia of 5000 m in a hypobaric chamber for 6 h/day. Barometric pressure (pB) was lowered to the level equivalent to an altitude of 5000 m (pB=54 kPa; po2=11.3 kPa). The total number of exposures was 42-day. The temperature in the chamber was maintained at 22–24 oC. The animals were examined the day after the last hypoxic exposure. The control group of animals was kept under normoxic environmental conditions. All animals were maintained with a natural light-dark cycle (12 h Light: 12 h Dark).
Isolated rat heart perfusion The rats were anesthetized with sodium pentobarbital (60 mg/kg, ip) as previously described [9]. Hearts were quickly excised and mounted on a Langendorff apparatus for a retrograde perfusion with Krebs–Henseleit solution (K-H buffer solution) at a constant pressure of 80 mmHg. K-H buffer solution contains (mmol/L): NaCl 118.0, KCl 4.7, CaCl2 2.5, MgSO4 1.2, NaHCO3 25.0, KH2PO4 1.2, glucose 11.0, and sodium pyruvate 2.0. The medium was continuously gassed with 95% O2 and 5% CO2 (pH 7.4) and maintained at 37 oC. A water-filled latex balloon connected to a pressure transducer (Gould P23Db) was introduced into the left ventricle via the mitral valve to record isovolumic left ventricular pressure. The balloon volume was adjusted to achieve a stable left ventricular end-diastolic pressure (LVEDP) of 5–10 mmHg during the initial equilibration. Heart rate (HR), left ventricular peak systolic pressure (LVPSP), LVEDP, left ventricular developed pressure (LVDP), coronary flow (CF), and the peak rate of pressure developed (±dp/dtmax) were monitored using a PowerLab system (AD Instrument Ltd, Castle Hill, Australia). LVDP=LVPSP–LVEDP. Pressure-rate product (PRP) was calculated, PRP = HR×LVDP.
Experimental protocol and groups We chose AG, an iNOS-specific inhibitor[18], to detect its effect on the cardioprotection of IH. Rats were divided into four groups: (1) corresponding control (CON) group; (2) CON+AG group; (3) IH group; and (4) IH+AG group. In the present study, we used 20 min baseline perfusion, 30 min no-flow global ischemia, followed by 30 min reperfusion protocol. The hearts of rats in the drug group were treated with AG 100 µmol/L for 5 min before ischemia and maintained during 30 min ischemia, followed by 30 min reperfusion with K-H buffer solution. To obtain samples for biochemical, RT-PCR, and Western-blot experiments, each group was further divided into three subgroups: (1) baseline perfusion group: 20 min baseline perfusion; (2) ischemia group: 20 min baseline perfusion followed by 30 min no-flow global ischemia; and (3) ischemia/reperfusion group: 20 min baseline perfusion, 30 min no-flow global ischemia, followed by 30 min reperfusion. At the end of baseline perfusion, ischemia, and reperfusion, respec-tively, the hearts were dissected into left and right ventricles, frozen in liquid nitrogen, and stored at –80 oC.
Nitrate plus nitrite (NOx) measurement NOx, the stable end product of NO, was assessed as nitrite concentration after conversion of nitrate to nitrite with nitrate reductase and measured using a commercial kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). NOx was designated as μmol/g protein. Its concentration was determined at an optical density of 550 nm in a spectrophotometric method. Protein determination was carried out according to the Bradford method using bovine serum albumin as the standard.
RNA isolation and semi-quantitative determination of iNOS using RT-PCR Total RNA was extracted from frozen ventricular tissue with Trizol reagent and quantified by absorption at 260 nm. Reverse transcription from 0.5 µg total RNA was incubated in a 20-µL mixture containing 40 U reverse transcriptase, 500 µmol/L of each dNTP, 500 ng oligo(dT) and reaction buffer at 42 °C for 50 min. The sequences of the iNOS primers were 5'-ACTGCTGGTGGTGA-CAAG-3' (forward) and 5'-CGTTGGAAGTGTA-GCGTT-3' (reverse), allowing the amplification of a 333-bp fragment; the sequences of the M28S primers were 5' AGCAGCCGA-CTTAGAACTGG-3' (forward) and 5'-TAGGGACAGTGGGA-ATCTCG-3' (reverse), allowing the amplification of a 250-bp fragment. The PCR contained 0.1 µmol/L of each primer, 200 µmol/L of each dNTP, 2.5 mmol/L MgCl2 reaction buffer and 1 U Taq DNA polymerase in a final reaction volume of 20 µL. The reaction mixture was incubated in a thermocycler (Eppendorf Mastercycler gradient, Germany) programmed to predenature at 94 °C for 5 min, denature at 94 °C for 40 s, anneal at 60 °C for 40 s, and extend at 72 °C for 40 s for a total of 38 cycles. The last cycle was followed by a final elongation at 72 °C for 5 min and cooled to 4 °C. A pilot experiment had shown that this cycle number allowed product detection within the linear phase of amplification.
The amplified products were electrophoresced on a 1.0% agarose gel stained with ethidium bromide, visualized under ultraviolet light, and scanned using Gel doc 2000 (Bio-Rad, Richmond, CA, USA). The results were expressed as the relative intensity of bands for iNOS PCR product normalized by the intensity of the band for M28S.
Preparation of protein extracts and Western blot analysis of iNOS isozyme The protein extracts were prepared by homogenizing the left ventricles in isotonic sucrose buffer A (mmol/L): Tris-HCl 20.0, sucrose 250.0, Na3VO4 0.03, MgCl2 2.0, edetic acid 2.0, egtazic acid 0.5, PMSF 2.0, DTT 1.0, protease inhibitor cocktail 0.02% (v/v), pH 7.4. The homogenates were centrifuged at 100 000×g for 60 min at 4 °C to separate the particulate fraction from the cytosolic fraction. The supernatant containing soluble NOS, designated as cytosol, was used for the Western blot analysis of iNOS.
After boiling for 10 min, equivalent amounts of cytosolic protein (40 µg) were separated by 8% denaturing sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), electroblotted onto a nitrocellulose membrane and immunoreacted with iNOS primary antibody (Santa Cruz Biotech, Santa Cruz, CA, USA) overnight at 4 °C, followed by a 2-h incubation at room temperature with the second antibody (goat anti-rabbit IgG, Sigma Chemical Company, St Louis, MO, USA) conjugated with horseradish peroxidase. The iNOS isozyme was detected using enhanced chemiluminescence (ECL, Amersham Biosciences, UK) as 130-kDa bands. Each iNOS isozyme signal was normalized to the standard signal of the normoxic control heart. The scanned image was imported into Adobe Photoshop software; scanning densitometry was used for quantitative analysis of the data.
Materials and reagents AG was purchased from the Sigma Chemical Company; Trizol isolation reagent was from Invitrogen Life Technologies (San Diego, CA, USA); primers of iNOS and M28S were synthesized by Sangon Bioengineering Company (Shanghai, China); and the reverse transcription system was obtained from Promega (Madison, WI, USA).
Statistical analysis All data are expressed as mean±SD. Statistical analysis were carried out using one-way ANOVA or Student’s t tests when appropriate. Differences were considered significant at P<0.05.
Results
Effects of AG on recovery of CF and ventricular function after I/R in IH and normoxic rats Our previous study showed that the ratio of whole ventricle weight to body weight of rats in IH groups was not significantly different from normoxic control animals, which meant that intermittent hypoxia in this experimental condition did not result in heart hypertrophy[6,19]. The present study demonstrated that CF was slightly but significantly higher in IH hearts during baseline perfusion (Table 1). CF dramatically decreased during reperfusion in all groups, but the improvement of CF during reperfusion was greater in the IH group compared with the normoxic control group. The addition of AG 100 µmol/L did not significantly change either pre-ischemic CF or LVDP, but inhibited pre-ischemic ±dp/dtmax in normoxic and IH hearts (data not shown). The addition of AG 100 µmol/L inhibited the improvement of CF during reperfusion in the IH group, but had no influence on normoxic hearts.
As shown in Table 1, HR did not change in any group and was not affected by AG 100 µmol/L. Baseline values of ventricular function variables did not differ in any group during baseline perfusion, except for CF, but the values of LVPSP, ±dp/dtmax, and PRP were greatly decreased, whereas LVEDP significantly increased during reperfusion. During reperfusion, values of ±dp/dtmax, LVEDP, and PRP in the IH group were superior to the normoxic group, suggesting that left ventricular functional recovery was modestly facilitated by IH adaptation. AG significantly inhibited the recovery of IH hearts, whereas it had no effect on normoxic hearts during reperfusion (Figure 1).
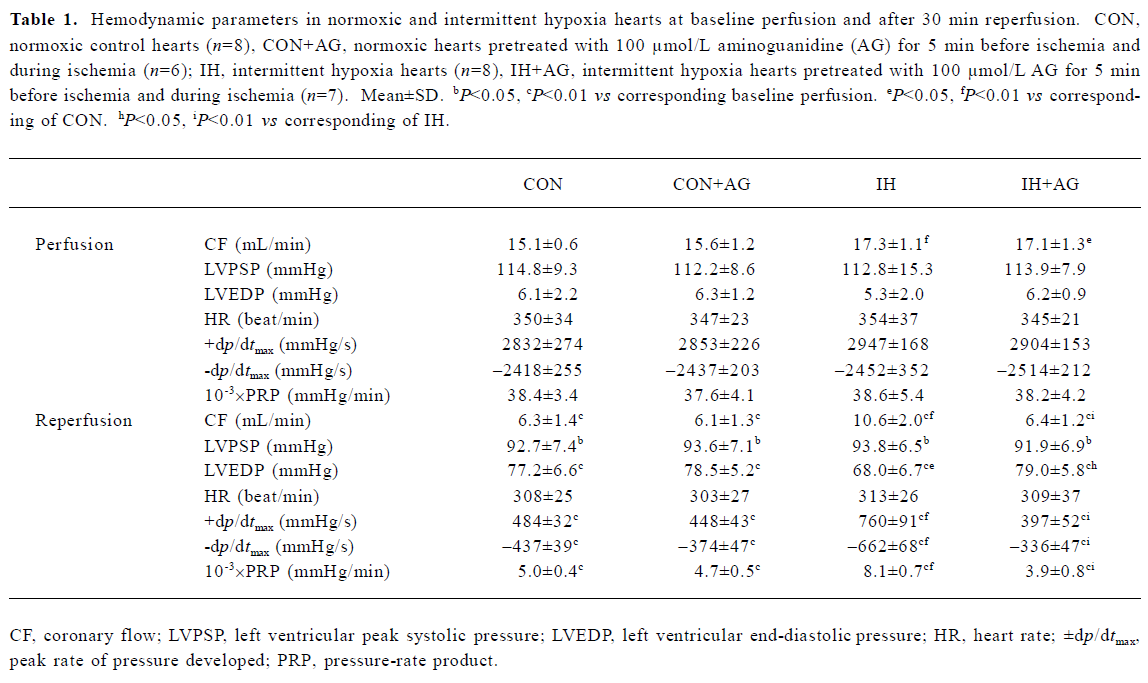
Full table
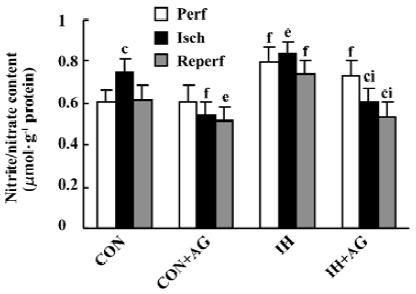
Effects of AG on the nitrate/nitrite content of IH and normoxic rat hearts The biochemical experiment demonstrated that the baseline content of NOx in IH hearts was 30.4% higher than that in normoxic hearts (P<0.01). After 30 min ischemia, the NOx level in normoxic heart tissue increased from 0.608±0.060 to 0.747±0.062 µmol/g protein, whereas in IH heart tissue there was no significant change (P>0.05, 0.793±0.075 vs 0.837±0.054 µmol/g protein). The addition of AG 100 µmol/L significantly diminished the content of NOx in IH hearts from 0.837±0.054 to 0.606±0.070 µmol/g protein and from 0.747±0.062 to 0.541±0.062 µmol/g protein in normoxic hearts during the ischemia period. AG also significantly decreased the content of NOx in IH hearts from 0.742±0.062 to 0.535±0.072 µmol/g protein and from 0.617±0.072 to 0.519±0.059 µmol/g protein in normoxic hearts during the reperfusion period (Figure 2).
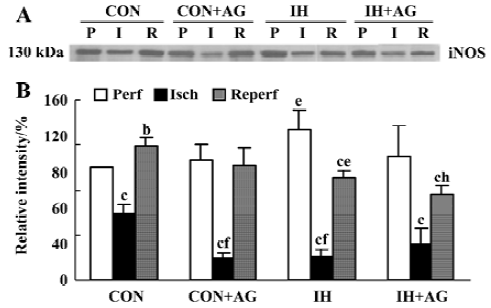
Effects of AG on iNOS mRNA expression in IH and normoxic rat hearts The expression of iNOS mRNA is shown in Figure 3A,3B. The baseline level of iNOS mRNA in IH hearts was higher than that in normoxic hearts by 50.2% (P<0.01). After reperfusion, iNOS mRNA level in normoxic hearts increased from 0.470±0.051 to 0.590±0.092 (P<0.05); however, the iNOS mRNA level in IH hearts decreased from 0.706±0.061 to 0.549±0.066 after reperfusion (P<0.05). The addition of 100 µmol/L AG significantly diminished iNOS mRNA level in normoxic and IH hearts after reperfusion; however, the reduced extent of IH hearts was higher than that of normoxic hearts (P<0.01).
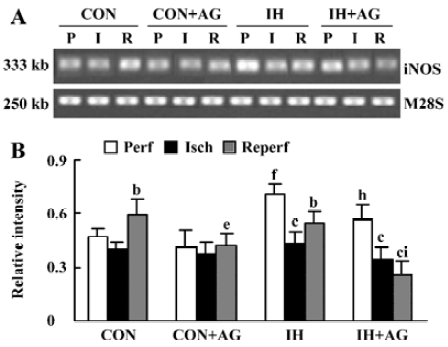
Effects of AG on iNOS protein expression in IH and normoxic rat hearts The expression of iNOS protein is demonstrated in Figure 4A, 4B. In accordance with the changes at mRNA level, the baseline level of iNOS protein in IH hearts was 33.8% higher than that of normoxic hearts (P<0.05). After reperfusion, iNOS protein level in the normoxic hearts increased by 18.4% (from 100% ± 0.0% to 118.4%±8.4%, P<0.05); however, iNOS protein level in IH hearts decreased by 31.9% (from 133.8%±16.6% to 91.1%±7.0%, P<0.01). After reperfusion, the level of iNOS protein between normoxic and IH hearts was significantly different (P<0.05). The addition of 100 µmol/L AG significantly decreased iNOS protein level in IH hearts; however, AG had no influence on iNOS protein expression in normoxic hearts after reperfusion. Ischemia significantly decreased iNOS protein expression in the different groups (P<0.05).
Discussion
The present study showed that IH increased the tolerance of hearts to I/R injury, determined by improved recovery of post-ischemic ventricular function. This protective effect was abolished by AG, suggesting that iNOS-derived NO may participate in the cardioprotection of IH. Our results also showed that the baseline level and the recovery of CF after ischemia were higher in IH hearts compared with normoxic hearts. Zhong et al’s study revealed that capillary densities were increased in IH hearts, which contributed to better functional recovery when isolated rat hearts were subjected to an I/R injury[6]. The recovery of post-ischemic CF in IH hearts was abolished by AG, suggesting that iNOS-derived NO may be involved in the improvement of CF by IH.
The level of NOS expression relates directly to the quantity of NOx production. To examine the effect of IH on iNOS expression, we investigated NOx content, iNOS mRNA, and protein expression in normoxic and IH rat hearts during baseline perfusion. Providing important additional insights into IH against I/R injury, we also examined NOx content, iNOS mRNA and protein expression in IH and normoxic rat hearts subjected to ischemia/reperfusion. This may help us to understand why IH could improve the post-ischemic recovery of left ventricular function. Our results showed that iNOS mRNA and protein were unregulated and that NOx content increased after IH adaptation in rat hearts. Palmer et al showed that hypoxia induced iNOS expression in cardiac myocytes and vascular endothelial cells[20,21]. Rouet-Benzineb et al also indicated that after 15 days of hypoxia there was a two-fold increase in iNOS protein abundance in rat left ventricles[22]. However, Baker et al demonstrated that increased tolerance to ischemia in rabbit hearts adapted to chronic hypoxia was associated with increased expression of eNOS isozymes, which led to an increase in myocardial nitrite/nitrate content and cGMP level[23]. The signaling functions of NO begin with its binding to protein receptors, such as guanylyl cyclase, which generates cGMP. In the present study, the elevated expression of myocardial iNOS in IH hearts compared to control hearts was also consistent with Zhong et al’s report[6], which showed that the myocardial cGMP level in IH rats increased compared to normoxia control rats. Baker et al suggested that a small increase in NO levels appears to be cardioprotective, whereas a large increase in NO production may be detrimental, resulting in vasodilation, decreased blood pressure and, perhaps, vascular leakage[23]. Importantly, in our study the magnitude of iNOS upregulation caused by IH was mild. This quantitative induction of iNOS may be critical in explaining the protective effects of iNOS following IH adaptation. The discrepancies between Baker et al’s study and ours lie in the age of the animals (adult vs neonatal), the species (rabbit vs rat), the experimental model, and the training duration and/or intensity.
In the present study, iNOS-derived NO was assessed by measuring the AG-inhibitable nitrite/nitrate content in IH and normoxia rat hearts. The addition of 100 µmol/L AG significantly decreased NOx content, iNOS mRNA and protein levels in IH hearts after reperfusion and abolished the protective effect of IH; AG also decreased NOx content and iNOS mRNA expression in normoxic hearts; however, the inhibitory extent was lower in normoxic hearts compared with IH hearts. This result suggests that iNOS is tonically higher in IH hearts compared with normoxic hearts and that iNOS may play an important role in the cardioprotection of IH.
Studies examining the role of NO in modulating ischemia/reperfusion injury are complicated. There are conflicting reports about the changes in NO content during ischemia/reperfusion period. A number of studies have demonstrated that NO content increased in hearts subjected to ischemia[24,25]. Studies have also shown that short-term ischemia leads to an increase in iNOS activity and expression; however, iNOS activity and expression decreased with prolonged ischemia. This may be related to a deficiency in essential co-factors for protein synthesis and stability (eg, haem, FAD, FMN, calmodulin). In addition to NO production by specific NOS, Zweier et al also demonstrated that NO could be generated in tissues by either direct disproportionation or reduction of nitrite to NO under the acidic and highly reduced conditions that occur in disease states, such as ischemia[26]. Thus, we considered that the basal level of nitrite and nitrate was a better index of nitric oxide production from the aerobically perfused heart.
In addition, we found that iNOS mRNA and protein level in normoxic hearts increased after reperfusion; however, iNOS mRNA and protein level decreased in IH hearts. The results from Zingarelli et al’s study differed from our result in that iNOS mRNA in wild-type mice was significantly increased after 30 min reperfusion compared to the basal level [27]. In the present study, the level of iNOS protein in normoxic hearts was higher than the level in IH hearts after I/R. It is known that induction of high-output iNOS usually occurs in an oxidative environment, and thus high levels of NO have the opportunity to react with superoxide anion (O2–) leading to peroxynitrite (ONOO–) formation and cell toxicity[28]. The generation of ONOO– can account for the toxicity of NO in biological systems. Yasmin et al and Wang and Zweier’s studies indicated that generation of ONOO– at reperfusion contributed to the I/R injury in isolated rat hearts[29,30]. In general, O2– formation increases during the early period of reperfusion and reacts with NO to form ONOO–, which results in amino acid nitration and cellular injury[30]. Our previous study suggested that an increase in antioxidant capacity might play an important role in the effect of IH reducing I/R injury[5]. Based on the above investigations, we propose that during reperfusion, normoxic hearts may be predisposed to produce more O2– and ONOO– than IH hearts, and thus produce more potent toxicity than IH hearts. Our results also suggest that during reperfusion after sustained ischemia, NO may have bidirectional effects on myocardium because of the coexistence of NO and O2–. AG significantly diminished NOx content, iNOS mRNA and protein level in IH hearts after reperfusion and led to the inhibitory effect of AG on the recovery of left ventricular function of IH rat hearts. As far as we know, NO is considered to play a pivotal role in numerous physiological and pathophysiological processes, with effects arising from both a lack and a surfeit of this chemically reactive molecule. This seemingly paradoxical behavior may be explained by the amount of NO generated, temporal-spatial intracellular compartmentalization of NO, and the intracellular redox environment.
The mechanisms leading to upregulation of iNOS after IH adaptation and the mechanism whereby iNOS-derived NO plays a role in the protection of IH have not been clarified and need to be further investigated. Mechanisms by which hypoxia induces gene expression include transcriptional and posttranscriptional regulation. The molecular mechanisms of hypoxia adaptations are centered on the activation of hypoxia-inducible factor 1 (HIF-1). During hypoxia, however, HIF-1α is stabilized, leading to accumulation of the active HIF-1α, HIF-1β heterodimer, which binds to specific recognition elements within promoter/enhancer regions of many target genes, the induction of which generates this cytoprotective process. The set of genes induced in this manner include iNOS[20,21], which drives cytoprotective events mediated by NO. At the same time, complex mechanisms may exist and interplay in the regulation of iNOS expression in the heart.
NO is an important endogenous regulatory molecule involved in a variety of biological functions. For example, it maintains coronary vasodilator tone, inhibits platelet aggregation and the adhesion of neutrophils to vascular endothelium[31]. It can also regulate myocardial contractile function. Studies have suggested that NO could also regulate oxygen demand and the delivery of oxygen[32]. Therefore, moderate increases in NO during intermittent hypoxia might be involved in the regulation of the above effects.
We concluded that IH upregulated the baseline level of iNOS mRNA and protein expression leading to an increase in NO production, which may play an important role in the cardiac protection of IH against I/R injury.
References
- Powell FL, Garcia N. Physiological effects of intermittent hypoxia. High Altitude Med Biol 2000;1:125-36.
- Prabhakar NR. Physiological and genomic consequences of intermittent hypoxia: invited review: oxygen sensing during intermittent hypoxia: cellular and molecular mechanisms. J Appl Physiol 2001;90:1986-94.
- Meerson FZ, Ustinova EE, Orlova EH. Prevention and elimination of heart arrhythmias by adaptation to intermittent high altitude hypoxia. Clin Cardiol 1987;10:783-9.
- Asemu G, Papousek F, Ostadal B, Kolar F. Adaptation to high altitude hypoxia protects the rat heart against ischemia-induced arrhythmias. Involvement of mitochondrial KATP channel. J Mol Cell Cardiol 1999;31:1821-31.
- Zhang Y, Zhong N, Zhu HF, Zhou ZN. Antiarrhythmic and antioxidative effects of intermittent hypoxia exposure on rat myocardium. Acta Physiol Sin 2000;52:89-92.
- Zhong N, Zhang Y, Zhu HF, Wang JC, Fang QZ, Zhou ZN. Myocardial capillary angiogenesis and coronary flow in ischemia tolerance rat by adaptation to intermittent high altitude hypoxia. Acta Pharmacol Sin 2002;23:305-10.
- Zhong N, Zhang Y, Fang QZ, Zhou ZN. Intermittent hypoxia exposure-induced heat-shock protein 70 expression increases resistance of rat heart to ischemic injury. Acta Pharmacol Sin 2000;21:467-72.
- Neckar J, Szarszoi O, Koten L, Papousek F, Ost'adal B, Grover GJ, et al. Effects of mitochondrial KATP modulators on cardioprotection induced by chronic high altitude hypoxia in rats. Cardiovasc Res 2002;55:567-75.
- Zhu HF, Dong JW, Zhu WZ, Ding HL, Zhou ZN. ATP-dependent potassium channels involved in the cardiac protection induced by intermittent hypoxia against ischemia/reperfusion injury. Life Sci 2003;73:1275-87.
- Dong JW, Zhu HF, Zhu WZ, Ding HL, Ma TM, Zhou ZN. Intermittent hypoxia attenuates ischemia/reperfusion induced apoptosis in cardiac myocytes via regulating Bcl-2/Bax expression. Cell Res 2003;13:385-91.
- Beall CM, Laskowski D, Strohl KP, Soria R, Villena M, Vargas E, et al. Pulmonary nitric oxide in mountain dwellers. Nature 2001;414:411-2.
- Jones SP, Girod WG, Palazzo AJ, Granger DN, Grisham MB, Jourd'Heuil D, et al. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am J Physiol 1999;276:H1567-73.
- Weiland U, Haendeler J, Ihling C, Albus U, Scholz W, Ruetten H, et al. Inhibition of endogenous nitric oxide synthase potentiates ischemia-reperfusion-induced myocardial apoptosis via a caspase-3 dependent pathway. Cardiovasc Res 2000;45:671-8.
- Brunner F, Maier R, Andrew P, Wolkart G, Zechner R, Mayer B. Attenuation of myocardial ischemia/reperfusion injury in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Cardiovasc Res 2003;57:55-62.
- Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, et al. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci USA 1999;96:11507-12.
- Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and precondi-tioning: an overview of a decade of research. J Mol Cell Cardiol 2001;33:1897-918.
- Neckar J, Papousek F, Novakova O, Ost’adal B, Kolar F. Cardio-protective effects of chronic hypoxia and ischaemic preconditioning are not additive. Basic Res Cardiol 2002;97:161-7.
- Misko TP, Moore WM, Kasten TP, Nickols GA, Corbett JA, Tilton RG, et al. Selective inhibition of the inducible nitric oxide synthase by aminoguanidine. Eur J Pharmacol 1993;233:119-25.
- Zhang Y, Zhong N, Zhou ZN. Effects of intermittent hypoxia on action potential and contraction on non-ischemic and ischemic rat papillary muscle. Life Sci 2000;67:2465-71.
- Palmer LA, Semenza GL, Stoler MH, Johns RA. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Physiol 1998;274:L212-19.
- Jung F, Palmer LA, Zhou N, Johns RA. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ Res 2000;86:319-25.
- Rouet-Benzineb P, Eddahibi S, Raffestin B, Laplace M, Depond S, Adnot S, et al. Induction of cardiac nitric oxide synthase 2 in rats exposed to chronic hypoxia. J Mol Cell Cardiol 1999;31:1697-708.
- Baker JE, Holman P, Kalyanaraman B, Griffith OW, Pritchard KA Jr. Adaptation to chronic hypoxia confers tolerance to subsequent myocardial ischemia by increased nitric oxide production. Ann N Y Acad Sci 1999;874:236-53.
- Zweier JL, Wang PH, Kuppusamy P. Direct measurement of nitric oxide generation in the ischemic heart using electron paramagnetic resonance spectroscopy. J Biol Chem 1995;270:304-7.
- Node K, Kitakaze M, Kosaka H, Komamura K, Minamino T, Inoue M, et al. Increased release of NO during ischemia reduces myocardial contractility and improves metabolic dysfunction. Circulation 1996;93:356-64.
- Zweier JL, Samouilov A, Kuppusamy P. Non-enzymatic nitric oxide synthesis in biological systems. Biochim Biophys Acta 1999;1411:250-62.
- Zingarelli B, Hake PW, Yang Z, O'Connor M, Denenberg A, Wong HR. Absence of inducible nitric oxide synthase modulates early reperfusion-induced NF-kappaB and AP-1 activation and enhances myocardial damage. FASEB J 2002;16:327-42.
- Xie Q, Nathan C. The high-output nitric oxide pathway: role and regulation. J Leukoc Biol 1994;56:576-82.
- Yasmin W, Strynadka KD, Schulz R. Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts. Cardiovasc Res 1997;33:422-32.
- Wang P, Zweier JL. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J Biol Chem 1996;271:29223-30.
- Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function. Circ Res 1996;79:363-80.
- Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta 1999;1411:351-69.