
Exo+ proofreading polymerases mediate genetic analysis and its application in biomedical studies1
High-fidelity DNA polymerases mediate geno-typing
3´Terminal-labeled primer extension Terminal-labeled primer extension is a single nucleotide polymorphism (SNP) assay consisting of 3´ terminal-labeled, allele-specific primers and DNA polymerases with proofreading activity. Both 3´ terminal [ 3H]-labeled and fluorescent-labeled primers have been successfully applied in genotyping analyses. The 3´ terminal mismatched nucleotide that bears the signal to be detected was removed by the proofreading function, whereas the label was retained when the primer and template were perfectly matched. The terminal-labeled primer extension approach has several advantages over current SNP assays. The most significant advantage is that it greatly decreases false positive results by a direct consequence of the proofreading activity of Exo+ polymerases. The second advantage of terminal-labeled primer extension is its high sensitivity. Terminally labeled primer extension harnesses the power of polymerase chain reaction (PCR) to improve the efficiency of genetic analysis[1–7].
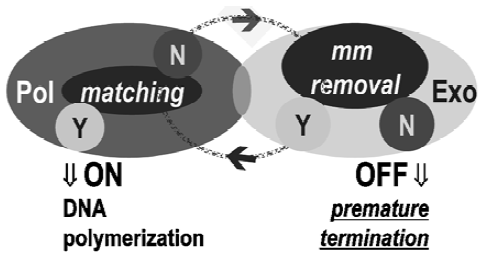
SNP-triggered on/off switch Exo+ polymerases together with 3´ phosphorothioate-modified mismatched primers work as an off switch in DNA polymerization. Phosphorothioate modification renders oligonucleotides nuclease–resistant, which blocks mismatch excision, a strategy widely used in antisense technology as well as in single base extension. For 3´ allele-specific primers with phosphorothioate modification, a perfectly matched primer turns on DNA polymerization, whereas mismatched primers turn it off, resulting in no product. This breakthrough observation of an on/off switch action has been repeatedly confirmed using either short artificial amplicons or natural genomic DNA templates. The off switch directly resulted from 3´exonuclease activity and has been well supported by comparisons of a variety of DNA polymerases in both linear and exponential amplification with phosphorothioate-modified, allele-specific primers[3–13].
The crucial structural components of the on/off switch are: (i) allele-specific primers with 3´ terminal exonuclease-resistant modification; and (ii) DNA polymerases possessing 3´ exonuclease activity. In recent studies by Di Giusto et al[6,7,14], four types of DNA polymerases, T4+, T7+, KF+ and Vent, were tested and similar off switches were observed when the polymerases were combined with 3´ phosphoro-thioate-modified primers. Based on the new model of this proofreading mechanism (Figure 1), polymerases with 3´ exonuclease function should have a higher base discrimination ability over exo-polymerases regardless of the properties of the substrates used. In addition to comparing nine different DNA polymerases, Di Giusto et al[6,7,14] evaluated the effect of dNTP(2´-deoxynucleotide-5´-triphosphate), ddNTP (2´,3´-dideoxynucleotide-5´-triphosphate), and acyNTP (acyclo-2´-deoxynucleotide-5´-triphosphate) on the accuracy of primer extension. The maintenance of high fidelity with ddNTP and acyNTP allows the Exo+ polymerases to be applied in both exponential and linear primer extensions in SNP analysis. The latter was tested using MALDI-TOF MS (Matrix Assisted Laser Desorption/Ionization Time of Flight Mass Spectrometry) and is compatible with many other detection formats.
SNP-triggered reversed on/off switch or off/on switch The SNP triggered reversed on/off switch works in a way complementary to proofreading 3´ exonuclease-resistant or 3´ labeled primers. With the introduction of inert allele-specific primers or proprimers, matched amplicons turn off and mismatched amplicons turn on DNA polymerization. Inert primers of a perfectly matched amplicon are not processed by the 3´Exo domain of the high-fidelity DNA polymerases. In this circumstance, inert primers remain inactive and no DNA polymerization occurs. In contrast, the inert primers of mismatched amplicons trigger the 3´exonuclease excision process by which the mismatched 3´ terminal is removed and subsequent processes activate the 3´ hydroxyl group for DNA polymerization. One of the benefits of the reversed on/off switch is that positive results can screen all three mismatched nucleotides other than the complementary one, and this is a powerful tool in mutation detection.
To date two types of 3´ dehydroxylated primers, 3´ phosphorylated and 3´ hydrogenized, have been evaluated for use in the SNP-triggered reversed on/off switch. The 3´ phosphorylated primers can always be extended regardless of whether low-fidelity or high-fidelity polymerases are used. Fortunately, the 3´ hydrogenized primer works well as an inert primer that can not be extended without activation through removal of the 3´ terminal nucleotide residue. The SNP-operated reversed on/off switch is not a simple candidate method in mutation detection. One particularly important feature of the previously developed on/off switch and this new reversed on/off switch is the identical reaction conditions of the two types of Exo+ DNA polymerases mediated by the primer extension. In large-scale SNP scanning, the application of two complementary assays within one platform, such as multi-well plates or microarrays, will help to minimize wrongly genotyped SNP sites resulting from special local sequence contents. With the application of the reversed on/off switch using the inert primers and the on/off switch using the 3´ exonuclease-resistant primers, their complementary effect will help to increase assay sensitivity and reliability in genetic analysis. The on/off switch provides assays for precise detection of the location and type of mutation, whereas the reversed on/off switch serves as a very powerful and efficient assay in unknown mutation scanning[11]. The reversed on/off switch was first described in 1998 as a proofreading PCR by Bi and Stambrook[15].
Genomapping applications other than SNP assay
The high-fidelity features of the Exo+ polymerases mediated on/off and off/on switch can be widely used in a variety of genetic analysis, other than SNP assays. These include both genotyping and gene expression profiling. Genotyping focuses on the sequencing context and gene expression profiling evaluates both the sequence context and the functional levels of any specific isoforms of a given gene product. The Exo+ polymerases mediated assays provide methods useful for these two types of genetic assays simultaneously, an advantage favor to pharmacogenetic studies. To simplify the terminology, we refer the combination of genotyping and gene expression profiling as genomapping analysis.
Analysis of mutations with short deletion/insertion Most of the currently available SNP assays, such as the single base extension assay, can not discriminate between wild-type alleles and mutants with small insertions or deletions. The aforementioned data illustrates the powerful mutation detection ability of the novel on/off switch, indicating its potential in the analysis of both SNP and other types of mutations. Except for the deletion or insertion of identical short repeats, the application of this on/off switch provides an efficient and high throughput compatible assay for mutation analysis[4–11,16].
Detection of rare alleles using the novel on/off switch The novel SNP-operated on/off switch is more sensitive than many conventional mutation analysis methods, including preferential amplification of the mutant allele, preferential destruction of the wild-type allele and spatial separation of mutant from wild-type alleles, by virtue of its use of Exo+ high-fidelity DNA polymerases. This breakthrough observation of an on/off switch action was repeatedly confirmed using either short artificial amplicons or natural genomic DNA templates[11]. Another outstanding feature of the novel on/off switch is its flexibility and its ability to apply double switches in a single reaction (for example both the forward and the reverse primers are 3´-phosphorothioate modified and allele specific). The double-switch approach does not compromise sensitivity as is the case in most other primer-extension-based SNP assays.
Allele frequency estimation in pooled DNA samples The application of pooled DNA samples in initial studies can help to identify significant divergence in allele frequencies between case and control populations for further, more extensive association studies or for haplotype analysis. Invader technology, Bi-PAP (double directions-pyrophos-phorolysis activated polymerization), and the SNP-operated on/off switch can all be used in allele frequency estimation. Among these three assays, the on/off switch has the advantage of simplicity, sensitivity, and accuracy. The application of the novel on/off switch in allele frequency estimation is expected to lower the cost and increase the accuracy of allele estimation using pooled DNA samples. Because the analysis of pooled samples requires only a handful of reactions per SNP, low assay development cost becomes of paramount importance.
High-fidelity gene expression profiling with the novel on/off switch
In our SNP and mutation detection assays, we have shown that the on/off switch can recognize a single mismatch in six nucleotides upstream of the 3´terminus. In most circumstances, Exo+ DNA polymerases can discriminate single mismatched nucleotides several bases upstream of the primer-3´-termini, but this varies according to the amplicons and the enzymes[3].
In the early stage of gene expression analysis, mRNA is the target material. Such methods include Northern blotting and ribonuclease protection assays. Although these methods could be compatible with high throughput scalability, the ribonuclease protection assay is still, to date, the most reliable method in gene expression profiling. Microarray technology has solved the high throughput issue for genome-wide gene expression profiling. However, cDNA is an indirect target because it cannot completely represent the type and abundance of mRNA. False positives are particularly high when post reverse transcription amplification by Taq polymerases is used. To minimize the errors introduced by Taq polymerase-mediated amplification, high-fidelity DNA polymerases have recently been used in genome-wide gene expression profiling. Because proofreading polymerases display up to two orders of magnitude better amplification fidelity, the application of high-fidelity polymerases has great potential in improving the reliability of microarray-based gene expression profiling.
In general, false positives in gene expression profiling arise from three sources: mismatched incorporation in reverse transcription, mismatched priming, and mismatched incorporation during primer extension. Although the high-fidelity gene expression assay described in the present study has nothing to do with the errors introduced from reverse transcription, it eliminated the mismatchedpriming error and greatly minimized the mismatched incorporation during primer extension. Increased fidelity in gene expression profiling not only benefits assay sensitivity and reliability, it also provides new applications in genetic analysis. The differential amplification of the on/off switch allows for the profiling of very rare abundant transcripts in normal tissues or cancer tissues. It also offers simple and convenient methods for comparing isoforms, inter-species or intra-species.
Suggested strategies for somatic mutation load assay
It was almost impossible to analyze rare mutations or mutation loads before the development of highly sensitive genotyping methods. The false positives recorded in conventional assay methods, in both hybridization-based and enzyme-based assays, are higher than the mutation rate. Theoretically, our recently developed Exo+ polymerase-mediated on/off switch is a good method for examining rare mutations because it almost eliminates false positives. However, assay sensitivity is another obstacle facing the detection of rare mutations. For example, for single-copy genes, 1 mg genomic DNA only contains 3×105 molecules of relevant genes. Theoretically, to detect a single mutant out of 1×109–1×1010 wild-type molecules (based on the mutation rate in human somatic cells per generation), at least mg scale genomic DNA is needed. These results in two immediate problems: technical difficulties in obtaining the large amount of human genomic DNA required, and the inhibitory effect of large amounts of DNA on PCR reactions.
An alternative approach in somatic mutation load analysis is to use genetic elements with high-copy numbers in the genome. As spontaneous mutation rates vary among genes and tissues, interspersed repeats might be a better choice than tandem repeats. A combination of highly sensitive SNP genotyping methods and a high-copy number of interspersed repeats may provide a practical and useful strategy for somatic mutation load assays at the genome level.
Selection of interspersed repeats for somatic mutation load analysis Short, interspersed nucleotide element (SINE) repeats, such as Alu elements with a high incidence of CpG dinucleotides in new Alu inserts, predisposes a higher mutation rate than long, interspersed nucleotide elements (LINE). Approximately half of the SNP in young Alu elements fall in the region of CpG dinucleotides. For somatic mutation load analysis, it appears that the targeting nucleotides to be chosen need to be carefully sorted. If too many targets are CpG dinucleotides, the overall estimated somatic mutation load might be biased and higher than the actual load.
In the case of this example, 1 ng of genomic DNA used in a somatic mutation load assay with a high-copy number repeat sequence will have the same power as approximately 1 mg of genomic DNA when targeting single-copy genes. Another issue regarding our strategies for somatic mutation load analysis is validation using genomic DNA samples that have different somatic mutations as expected, such as DNA from cancer tissue versus DNA from normal tissue, or DNA from cells in their early passages versus that from late passages.
Alternative opportunities from mitochondrial DNA The large number of mitochondria present in every mammalian cell (1000–10 000) makes this extra-nuclear genetic material very useful in somatic mutation analyses. Furthermore, studies have reported that mitochondrial DNA (mtDNA) has a mutation rate of one base mutation per 1500–3000 years of evolution, which is believed to be a higher rate than chromosomal DNA. The existence of homoplasmy and heteroplasmy in mitochondria mutation and their close relationship with the development of disease strongly suggests the value of testing the mutation load of mtDNA in future studies of personalized medicine. Some mtDNA somatic mutation showed a dose-dependent pattern in causing diseases[17,18]
One technical advantage of sequence comparisons is the very-low copy numbers of well-matched regions between mitochondrial and chromosomal DNA. This is especially true for mitochondrial tRNA genes, which are never observed in more than five copies in chromosomal DNA. This feature allows somatic mutation load assays to be separately designed for chromosomal and mitochondrial DNA without preparing separate DNA samples.
Target simplification by using in vitro amplified DNA Genomic DNA can be easily simplified and amplified using routine PCR technology. From a technical point of view, a combination of the PCR amplification of target DNA and a subsequent mutation load detection using one of the mutation quantitative analysis methods outlined can be viewed as a type of nested PCR. An advantage of this nested strategy is that any chromosomal region can be used as the target in somatic mutation load analysis.
In addition, many types of RNA are transcribed at highly abundant levels. Therefore, cDNA obtained immediately from in vitro reverse transcription or with a low-cycle-number PCR amplification can be a useful target in somatic mutation load analysis. A potential drawback of using RNA as the target in somatic mutation load analyses is that most mutated RNA are not as stable as their wild-type partners, which may decrease the opportunity for accurately detecting mutations. Therefore, RNA can act as one of the targets, but can not be used as the sole target in practical somatic mutation load analyses when a global approximate of the mutation load is required.
Conclusion
Significant advances after the completion of the Human Genome Project, including bioinformatic algorithms and highly sensitive SNP genotyping technologies, have provided great possibilities for studies relating genotype with phenotype. High-fidelity DNA polymerases have been approved for use as new and very attractive members among a variety of enzymes in genetic analysis. The three types of new methods mediated by proofreading polymerases provide sensitive and reliable tools for genomapping analyses consisting of genotyping, gene expression profiling, and mutation load assay. Genotyping, including SNP screening, is now widely used in pharmacogenetic studies. The most immediate tasks in somatic mutation load analysis are the selection of the genetic sequence set as representative targets for mutation assay, and the validation of these representative sequences using one or more of the three mutation analysis methods discussed. It is reasonable to expect that somatic mutation load analysis, especially the mitochondrial somatic mutation load, will have an important impact on both basic research and clinical practice.
References
- Zhang J, Li K. The 3´ terminal labeled primer extension: a new method of high throughput screening for SNP analysis. Curr Drug Disc 2001;9:21-3.
- Zhang J, Li K, Deng Z, Liao D, Fang W, Zhang X. Efficient mutagenesis method for producing the template of single nucleotide polymorphisms. Mol Biotechnol 2003;24:105-10.
- Zhang J, Li K, Liao D, Pardinas JR, Chen L, Zhang X. Different application of polymerases with and without proofreading activity in single-nucleotide polymorphism analysis. Lab Invest 2003;83:1147-54.
- Li K, Zhang J. New SNP assays from an old concept of proofreading. Curr Drug Disc 2003;11:37-9.
- Cahill P, Bakis M, Hurley J, Kamath V, Nielsen W, Weymouth D, et al. Exo-proofreading, a versatile SNP scoring technology. Genome Res 2003;13:925-31.
- King GC, Di Giusto DA, Wlassoff WA, Giesebrecht S, Flening E, Tyrelle GD. Proofreading genotyping assays and electrochemical detection of SNPs. Hum Mutat 2004;23:420-5.
- Di Giusto DA, King GC. Strong positional preference in the interaction of LNA oligonucleotides with DNA polymerase and proofreading exonuclease activities: implications for genotyping assays. Nucleic Acids Res 2004;32:e32.
- Zhang J, Meng B, Liao D, Zhou L, Zhang X, Chen L, et al. De novo synthesis of PCR templates for the development of SARS diagnostic assay. Mol Biotechnol 2003;25:107-12.
- Zhang J, Li K. Single base discrimination mediated by proofreading 3´ phosphorothioate-modified primers. Mol Biotechnol 2003;25:223-8.
- Zhang J, Li K. On/off regulation of 3´ exonuclease excision to DNA polymerization by exo+ polymerase. J Biochem Mol Biol 2003;36:525-8.
- Zhang J, Chen LL, Guo ZF, Peng CY, Liao DF, Li K. On/off switch mediated by exo+ polymerases: experimental analysis for its physiological and technological implications. J Biochem Mol Biol 2003;36:529-32.
- Zhang J, Li K, Pardinas JR, Liao DF, Li HJ, Zhang X. SNP discrimination through proofreading and off-switch of Exo+ polymerase. Mol Biotechnol 2004;27:75-80.
- Gale JM, Tafoya GB. Evaluation of 15 polymerases and phosphorothioate primer modification for detection of UV-induced C:G to T:A mutations by allele-specific PCR. Photochem Photobiol 2004;79:461-9.
- Di Giusto DA, King GC. Single base extension with proofreading polymerases and phosphorothioate primers: improved fidelity in single-substrate assays. Nucleic Acid Res 2003;31:7.
- Bi W, Stambrook PJ. Detection of known mutation by proof-reading PCR. Nucleic Acids Res 1998;26:3073-5.
- Zhang J, Li K. New performance from an old member: SNP assay and de novo sequencing mediated by Exo+ DNA polymerase. J Biochem Mol Biol 2004;37:269-74.
- Li K, Chen LL, Peng CY, Wang C, Liao DF, Zhang J. Transgenic human assay and evolutionary role of mtDNA by mitochromic analysis. J Nanhua Univ 2004;32:1-3.
- Li K, Liao DF, Zhang J, Chen LL, Peng CY, Wang C. Mitochromics: a simplified transgenic human assay. Chin J Arterioscler 2003;11:601-2.