
Historic perspectives and recent advances in major animal models of hypertension1
Introduction
Hypertension is the prevalent cause of cardiovascular disease that leads to heart failure, stroke, renal failure, and, ultimately, to death. In the USA, approximately 50 million people have hypertension, according to Phase I estimates of the National Health and Nutrition Examination Survey III (NHANES III), from 1988 to 1991[1]. The prevalence is about 20%. According to a report[2], about 40% of white people and more than 50% of black people aged 65 or older suffer from some form of high blood pressure in the USA. Epidemiological surveys have demonstrated an increasing prevalence of hypertension in China, from 5.11% in 1958 to 7.73% in 1980 and 11.89% in 1991[3,4]. Although this incidence is lower than that in many developed countries such as the USA (20%)[1] and Canada[5], it represents 50 million people in 1980 and 90 million people in 1991 due to the size of the Chinese population. According to relevant reports[4,6], cardiovascular diseases are the leading causes of death in Chinese society.
The syndrome of hypertension is more than just an elevation of arterial pressure, although it is this aspect that the general public is most familiar with. Hypertension when fully developed is characterized by an increase in vascular resistance to blood flow, cardiac hypertrophy, often an increase in cardiac output, an increased output of sympathetic nervous system (SNS), changes within the vascular smooth muscles, often atherosclerosis, and abnormalities in renal function. Increased vascular resistance is due to constriction of the vascular smooth muscle, primarily in the high resistance vessels. The etiology of essential hypertension is not known and the causes of it may be multifactorial. It is generally believed that both genetic and environmental factors and their interactions play a critical role in the pathogenesis of hypertension and other cardiovascular diseases. Since hypertension and associated cardiovascular diseases are the leading causes of death of human beings, medical scientists are dedicated to elucidate the mechanism of and explore the treatment for hypertension.
It is noticed that animal models of human disease have been widely used to study etiology and pathogenesis of human disease, to prevent disease or to find a therapy and identify risk factors contributing to the disease. This literature reviews briefly advances in the most widely used rodent models of hypertension, including advantages and limitations. Hopefully, this will be useful to those in choosing appropriate animal models of hypertension for their researches. This comprehensive review may help physicians and scientists understand the pathogenesis of hypertension and therefore optimize therapeutic approaches. It is our expectation that the creation and utilization of novel animal models of hypertension will fundamentally advance the fields of cardiovascular medicine and physiology.
The major animal models of hypertension
Basically, animal models of hypertension comprise primary and secondary hypertension according to hypertension etiology (Figure 1). The primary hypertension includes genetically-induced and environmentally-induced hypertension and the secondary hypertension includes pharmacologically-induced and renal-induced hypertension by the way of induction.
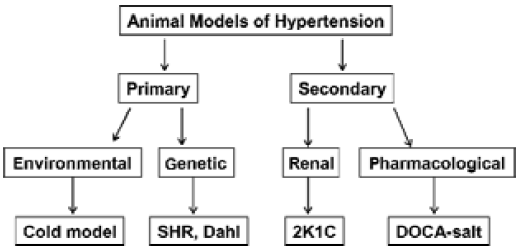
Genetically-induced hypertension Animals that have undergone artificial genetic manipulation are predestined to become hypertensive. Some investigators believe that genetically hypertensive rats comprise the most popular mo-dels to study essential hypertension[7]. One example is the spontaneously hypertensive rat (SHR), originally inbred from Wistar stock by Okamoto and Aoki, and their WKY inbred nonhypertensive controls. These rats develop hypertension at about 4–6 weeks of age, largely independent of die-tary levels of either Na+ or Cl-. Another model is the Dahl salt-sensitive rats, originally derived from Sprague-Dawley stock by Dahl on the basis of developing hypertension with high NaCl diet. When fed with normal salt diets, these rats become hypertensive, indicating that this is a genetic model of hypertension with the feature of salt sensitivity. O’Dowd and Rapp[8] have refined the genetic background of the original Dahl stock, and their salt-sensitive/Jr line is now preferred (along with their salt-resistant/Jr controls). Genetic analysis revealed linkage with loci close to the angiotensin converting enzyme (ACE) and guanylyl cyclase A (GCA)/atrial natriuretic factor (ANF) receptor genes in Dahl salt-sensitive rats[9]. It was found that certain alleles at the GCA and ACE loci (or at loci closely linked to them) had a significant impact on blood pressure response to high salt in Dahl salt-sensitive rats.
The SHR was generated by inbreeding Wistar rats with the highest blood pressure[7]. The SHR develop many features of hypertensive end-organ damages including cardiac hypertrophy, heart failure, and renal dysfunction. According to relevant reports[11,12], the SHR is the most commonly used genetic model although the mechanisms for producing the hypertension are not clear. Overactivity of the brain renin-angiotensin system (RAS) appears to be involved in initiating the development of spontaneous hypertension based on the finding that central injections of angiotensin II (AngII) receptor antagonists cause greater reductions in systolic blood pressure in SHR than in normotensives[13,14]. However, DePasquale and colleagues[15] reported that central administration of AT1 receptor blocker losartan (DuP 537) did not lower blood pressure in SHR. Thus, the above hypothesis needs further confirmation. Interestingly, some hemodynamic alterations which occur during the development of the disease are similar to human essential hypertension (ie, a high cardiac output early, and in the adult, a normal cardiac output and an increased vascular resistance)[16]. In this model, there is also an increase in the activity of the sympathetic nervous system as evidenced by increased renal sympathetic activity[16]. The renal blood flow and glomerular filtration rate are normal[17] and renal vascular resistance is increased[22]. The SHR stroke prone (SHR-SP) is a further developed sub-strain with even higher levels of blood pressure and a strong tendency to die from stroke[18]. The SHR has been used to evaluate genetic factors involved in hypertension. Although a wide variety of genes seems to cosegregate in various crosses[12], neither of these genes is confirmed to be the etiological gene for hypertension. The advantage of this model is that its pathophysiological changes are similar to those found in human essential hypertension[16,19]. A major drawback of this model is the lack of an appropriate control, which has been genetically altered yet is free from the disease and the complexity of the genetic mutations which have affected not only blood pressure but many other regulatory systems as well.
Transgenic hypertension models can be generated by over-expressing a specific gene. This type of genetic model usually has a relatively good normotensive control, the same strain of animals without genetic alteration. It is an excellent model to study the role of a specific gene in the pathogenesis of hypertension. However, the transgenic model may not mimic human essential hypertension as well as the SHR does in terms of pathophysiology. A representative of this type of hypertension is the TGR (mREN2)27 transgenic rats[10]. In this genetic model, the introduction and overexpression of the mouse Ren-2 gene in Sprague-Dawley rats leads to severe hypertension[10]. The appropriate control is the age-, body weight-, and sex-matched normotensive Sprague-Dawley rats without renin transgene.
Environmentally-induced hypertension In recent years, the important role of environmental factors in the disease process has received great attention from scientists and physicians. The primary interest is focused on the area of cardiovascular disorders, particularly hypertension. To understand better the ways in which the environmental factors affect the cardiovascular functions, researchers have tried to produce experimental hypertension in animals by exposing them to different experimental paradigms.
Stress-induced hypertension Epidemiologists and clinicians have long suspected that stressful life events can be a sufficient trigger for the expression of hypertension in some individuals[20]. Attempts to induce hypertension in animals using sensory stimulation have generally employed loud noises, flashing lights, and oscillating cages, either separately or together. For example, in one early study rats were subjected to 20 weeks of randomized flashing lights, motion, and loud noises[21]. After 12 weeks on this schedule, systolic blood pressure (SBP) values reached approximately 150–160 mmHg. Perhaps a better example of this type of experiment is found in a study by Lin and Li[22]. Stressed rats developed hypertension within two weeks after irregular foot electric-shocks combined with buzzing noise. It is accepted that activation of sympathetic nervous system[23] and the RAS[24] play a role in the initiation of stress-induced hypertension. Li[25] found that increased activity of the cholinergic system in the rostral ventrolateral medulla (rVLM) was also involved in the development of stress-induced hypertension. Plasma catecholamine, corticosterone, angiotensin II, glucose, and lipids were found to be increased during stress. In this model, about 40%–50% of stressed rats failed to develop hypertension[26].
Diet-induced hypertension It is known that long-term exposure to a special diet (high salt, fat, or sugar) results in dietary hypertension in some animals or humans. For example, chronic fructose treatment in rats repeatedly has been shown to elevate blood pressure associated with insulin resistance and hyperinsulinemia[27,28]. Hyperactivity of the SNS and the RAS, vascular hypertrophy, and sodium retention by the kidney tubules have been proposed to be some of the mechanisms by which fructose induces hypertension. By degeneration of capsaicin-sensitive nerves, Wang and colleagues[29,30] developed a salt-sensitive model of hypertension. In this model, the increased salt sensitivity is mediated by the enhancement of the SNS[29].
Cold-induced hypertension: Fregly[31] and Sun[32–36] found that chronic exposure of rats to mild cold (41 °F or 5 °C) induced hypertension including cardiac hypertrophy within 3 weeks. This is presently the only “naturally-occurring” form of experimentally-induced hypertension that is induced without surgical intervention, administration of excessive doses of drugs or hormones, or genetic manipulation. It is interesting that the elevated blood pressure of rats after 7 weeks of exposure to cold does not return to pre-cold exposure level during the 4 weeks after removal from cold. Thus, an elevation of blood pressure induced by a longer period of cold exposure might not be reversible after return to thermoneutral temperature. Intermittent exposure of rats to cold also induces hypertension, with a sigmoid relationship between the hours per day exposed to cold and systolic blood pressure.
Cold-induced hypertension (CIH) is also demonstrable in humans. Epidemiological surveys and clinical observations have established that people who live and work in cold areas have a high incidence of hypertension and related cardiovascular diseases[37–39]. Cold temperature (weather) makes hypertension more severe in hypertensive patients[39,40]. The cold winter season has the highest mortality and morbidity of cardiovascular diseases in a year[39–42]. Donaldson[38], after studying the relationship between outdoor temperature and blood pressure in men in central London between 1986 and 1992, reported that cold exposure of normal life in winter is sufficient to induce significant and prolonged hypertension in the general population.
It was originally assumed that cold-induced elevation of blood pressure was a direct vasoconstrictive effect of sympathetic nervous system (SNS). Indeed, the SNS is activated by chronic cold exposure and the plasma and urine levels of catecholamines are increased significantly in cold-exposed rats[32–34,43]. However, the in vitro vascular contractile response to α1-adrenoceptor agonist, phenylephrine, is decreased significantly in cold-exposed rats[44]. The in vivo pressor response to phenylephrine is significantly reduced by chronic cold exposure[45]. Thus, the vascular α1-adreno-ceptors are down-regulated during cold exposure. Several studies[32,34,36,46] have shown that blockade of the RAS at different sites could attenuate or prevent the cold-induced elevation of blood pressure. Antisense oligodeoxynucleo-tides to angiotensinogen mRNA or AT1-receptor mRNA reduce elevated blood pressure of cold-exposed rats[47]. Angio-tensinogen gene knockout delays and attenuates CIH[48]. It has been therefore suggested elsewhere[32–34,36,48] that the hyperactivity of the SNS initiates CIH probably via activation of the RAS. Indeed, plasma renin activity (PRA) is increased during exposure to cold and abolishment of the rise in PRA by renal denervation prevents the development of CIH[32]. Cold-exposed rats had an elevation of blood pressure that was proportional to the concentration of NaCl in the diet[49], suggesting that CIH is sodium-dependent hypertension.
CIH is a prototypic model of environmentally-induced hypertension, which is similar in many ways to human hypertension. Induction of CIH requires a climate-controlled chamber, with a strict requirement of temperature and humidity. Perhaps, this is the limitation of this model.
Pharmacologically-induced hypertension The DOCA-salt-induced model of hypertension is a typical representative of pharmacologically-induced hypertension. A very high dose of deoxycorticosterone acetate (DOCA) ranging from about 300 to 1000 mg⋅kg-1⋅d-1 (sc) is required to induce hypertension in rats. Isotonic saline is the sole drinking fluid. This is an important co-factor because it expedites the deve-lopment of hypertension and makes it more severe. Thus, this model is salt-dependent in its initiation. It often needs surgical reduction of renal mass or unilateral nephrectomy. The combination of DOCA-salt and unilateral nephrectomy results in hypertension, cardiac and renal hypertrophy, and nephrosclerosis[50]. DOCA-salt hypertension is a low renin and volume overloaded form of hypertension. There is evidence that arginine vasopressin (AVP) plays a role in both the development and maintenance of DOCA-salt hypertension[51,52]. It is believed that AVP is involved as a vasopressor hormone in the pathogenesis of malignant DOCA-salt hypertension[53]. The SNS also appears to be involved in the development of DOCA-salt hypertension[54]. The increased activity of the SNS may affect renal function through the renal nerve, as denervation of the kidneys delays the onset and decreases the severity of the disease[54,55]. Further studies[56] indicated that AngII receptor binding sites were increased in the brain, suggesting an up-regulation of AngII receptors. However, AngI converting enzyme inhibitors and AngII receptor blocker are ineffective in reducing blood pressure in low-renin, DOCA-salt hypertension[57,58]. In contrast, aldosterone receptor blockers and diuretics are effective in reducing blood pressure in this model[59]. Most recent studies indicate that the endothelin system plays an important role in the pathogenesis of DOCA-salt hypertension[60–65]. Oxidative stress may also be involved in DOCA-salt hypertension[65,66]. The major limitations of the DOCA-salt model are: 1) the pharmacological (large) doses of drug required; 2) requirement for surgical reduction of renal mass; and 3) ingestion of a large amount of NaCl required. The major advantage of this model is the potential to investigate the role of sodium in the developmental stages of hyperten-sion.
Renal-induced hypertension The physiological function of the kidney includes maintenance of electrolyte and fluid balance and secretion of renin, an important component of the RAS. Thus, its involvement in the regulation of blood pressure and its important role in the development of hypertension are well accepted. Since 1934, when Goldblatt and his co-workers[67] induced an elevation of blood pressure by partial constriction of the renal artery of the dog, many renal-induced models of hypertension have been successfully established in rats, rabbits, sheep, and cats[68]. Generally, renal-induced experimental hypertension includes two-kidney Goldblatt hypertension (constriction of one renal artery while the contralateral kidney is left intact) and one-kidney Goldblatt hypertension (one renal artery is constricted and the contralateral kidney is removed).
In the rat, by clipping one renal artery (leaving the contralateral kidney untouched) with a clip which induced a severe hypertension, a biphasic course in the plasma renin activity was found. In the initial phase, sodium retention occurred and was associated with a transient increase in plasma renin activity returning to control levels within a week[69,70]. Thus, it has been suggested that the RAS plays a role in the development of two-kidney Goldblatt hypertension[69-71]. The sustained elevation of blood pressure may still be AngII-dependent since a sub-pressor dose of AngII would result in an elevation of blood pressure due to a shift in the dose-response curve of AngII[72]. This suggests, but does not prove, that AngII receptors are upregulated in this model of hypertension. The initial elevation of blood pressure in the one-kidney Goldblatt model is also AngII-mediated[73]. Due to the absence of the other normal kidney, no compensatory increase in sodium and water excretion can occur, and hence, fluid volume is retained[74]. This model is thus a sodium-fluid volume-dependent model. This would be an ideal model for studying the role of volume expansion in the development of hypertension.
During the early developmental stage of these two renal-dependent models, when the clip is removed, arterial blood pressure returns to normal[70,75]. Thus, renal-induced hypertension is reversible and reproducible. Furthermore, these models provide a unique opportunity to investigate the changes which occur specifically at the level of the kidney, as well as the role of the kidney in the long-term blood pressure control. If the reversal of hypertension is time-depen-dent, it would suggest that relevant changes, perhaps structural, have developed.
Clinical implications of animal models of hypertension
It should be mentioned that each of the above models of experimental hypertension studies a specific aspect of hypertension. Neither of them encompasses all traits of human essential hypertension. Due to the unknown etiologies of hypertension, the use of various research models, each of which induces the disease by a different mechanism yet with the same end result, is advantageous. By using different types of experimental hypertension, scientists could identify and evaluate potential risk factors contributing to hypertension and related cardiovascular diseases. This would allow new and effective measures to be adopted for preventions and therapies. Usually, animal studies are essential to the success of clinical trials. It is noted that remarkable advances in cardiovascular medicine have been originated from experimental hypertension. Therefore, animal models of hypertension are very important approaches to the study of human hypertension.
Physicians and scientists could learn the developmental process and pathogenesis of hypertension from these animal models that are designed to mimic those of human hypertension. The induction of hypertension in animals could help understand the etiological factors that may be involved in essential hypertension. For example, hypertension and cardiovascular diseases (CVD) may have genetic predeter-minants. There is a predisposition to hypertension and CVD in those patients with a positive family history. The notion of genetic predisposition for the development of hypertension has been strengthened by the development of genetic or spontaneous models of hypertension. The pathophysiological changes of spontaneous hypertension in animals are very similar to those of human essential hypertension. Also, transgenic models of hypertension induced by targeting different genes are excellent tools to study genes relevant to hypertension. An animal model of DOCA-induced hypertension is an ideal model for evaluating the role of sodium in hypertension. Studies from DOCA-induced hypertension have provided a strong basis for using spironolactone (aldosterone receptor blocker) to treat clinical hypertension. Because some special diets (high salt, fat, or sugar) could result in dietary hypertension, restriction of these diets has been suggested by clinicians for preventive and therapeutic purposes. The cause of hypertension varies with hypertensive patients. Thus, it is important to identify the potential cause of hypertension prior to treatment; which guarantees appropriate and effective treatment. For example, stenosis of the renal artery could cause hypertension as learned from an animal model of renal-induced hypertension. In this case, surgical correction of the stenosis of the renal artery may be more important than treatment with antihypertensive drugs. In addition, research findings from experimental hypertension may have clinical implications. For example, the RAS has been found to play a critical role in the pathogenesis of CIH in rats. This has led to a clinical trial which proves that the ACE inhibitor (captopril) or AT1 receptor blocker (losartan) can control hypertension appropriately and effectively in hypertensive patients in winter. This also could help to control the high incidence of myocardial infarction and stroke during the cold season. In contrast, the use of animal models of hypertension would allow scientists to test new pharmacological treatments and gene therapy for hypertension and related CVD. Thus, what is learned from animal hypertension may reveal new insights into both preventive strategies and optimization of therapeutic approaches. It should be emphasized that any experimental data from animal models of hypertension must be tested and further validated in humans before they can be applied in hypertensive patients.
References
- Burt VL, Whelton P, Roccela EJ, Brown C, Cutler JA, Higgins M, et al. Prevalence of hypertension in the US adult population. Results from the third national health and nutrition examination survey, 1988–1991. Hypertension 1995;25:305-13.
- National Heart, Lung, and Blood Institute. Age Page. High blood pressure: a common but controllable disorder. Bethesda, MA, USA. Department of Health and Human Services and National Institute of Health. 1991.
- Wu YK, Lu CQ, Gao RC, Yu JS, Liu GC. Nationwide hypertension screening in China during 1979–1980. Chin Med J 1982;335:101-5.
- PRC National Blood Pressure Survey Cooperative Group. Prevalence and development trends of hypertension in China. Chin J Hypertens 1996;3:7-13.
- Joffres MR, Hamet P, Rabkin SW, Gelskey D, Hogan K, Fodor G. Prevalence, control and awareness of high blood pressure among Canadian adults. Can Med Assoc J 1992;146:1997-2005.
- Hamet P, Gong LS. Chinese contribution to trials on antihypertensive therapy. J Hypertens 1996;14:S123-9.
- Yagil Y, Yagil C. Genetic models of hypertension in experimental animals. Exp Nephrol 2001;9:1-9.
- O’Dowd BF, Rapp JP. Heterogeneity of rennin alleles in outbred Dahl salt-sensitive (Brookhaven) rats. Hypertension 1991;18:9-11.
- Deng Y, Rapp JP. Cosegregation of blood pressure with angiotensin converting enzyme and arterial natriuretic peptide receptor genes using Dahl salt sensitive rats. Nat Genet 1992;1:267-72.
- Langheinrich M, Lee MA, Bohm M, Pinto YM, Ganten D, Paul M. The hypertensive Ren-2 transgenic rat TGR (mREN2)27 in hypertension research. Characteristics and functional aspects. Am J Hypertens 1996;9:506-12.
- Pravenec M, Zidek V, Landa V, Kostka V, Musilova A, Kazdova L, et al. Genetic analysis of cardiovascular risk factor clustering in spontaneous hypertension. Folia Biol Praha 2000;46:233-40.
- Yang RH, Jin H, Wyss JM, Oparil S. Depressor effect of blocking angiotensin subtype 1 receptors in the anterior hypothalamus. Hypertension 1992;19:475-81.
- Pinto YM, Paul M, Ganten D. Lessons from rat models of hypertension: from Goldblatt to genetic engineering. Cardiovasc Res 1975;39:77-88.
- Mann JF, Phillips MI, Dietz R, Haebara H, Ganten D. Effects of central and peripheral angiotensin blockade in hypertensive rats. Am J Physiol 1978;234:H629-37.
- DePasquale MJ, Fossa AA, Holt WF, Mangiapane ML. Central DuP 753 does not lower blood pressure in spontaneously hypertensive rats. Hypertension 1992;19:668-71.
- Trippodo NC, Frohlich ED. Similarities of genetic (spontaneous) hypertension: man and rat. Circ Res 1981;48:309-19.
- Arendshorst WJ, Beierwaltes D. Renal and nephron hemodynamics in spontaneously hypertensive rats. Am J Physiol 1979;236:F246-51.
- Yamori Y. Development of the spontaneously hypertensive rat (SHR) the stroke prone hypertensive SHR (SHRSP) and their various substrain models for hypertension-related cardiovascular diseases. In: Ganten D, de Jong W (volume editors) Birkenhager WH, Reid JT (series editors). Handbook of hypertension, v 16: Experimental and genetic models of hypertension. Amsterdam: Elsevier; 1994. p23–31.
- Coleman TG, Guyton AC, Young DB. The role of the kidney in essential hypertension. Clin Exp Pharmacol Physiol 1975;2:571-81.
- Zimmerman RS, Frohlich ED. Stress and hypertension. J Hyper-tens 1990;8:S103-7.
- Smookler HH, Buckley JP. Relationships between brain catecholamine synthesis, pituitary adrenal function and the production of hypertension during prolonged exposure to environmental stress. Int I Neuropharmacol 1969;8:33-41.
- Lin Q, Li P. The effects of chronic stress on blood pressure and heart rate in rats. Chin J Physiol Sci 1990;6:101-7.
- Smith PA, Graham LN, Mackintosh AF, Stoker JB, Mary DA. Sympathetic neural mechanisms in white-coat hypertension. J Am Coll Cardiol 2002;40:126-32.
- Coste SC, Brooks VL, McCarron DA, Hatton DC. Captopril and stress-induced hypertension in the borderline hypertensive rat. J Hypertens 1995;13:1391-8.
- Li P, Zhu DN, Kao KM, Lin Q, Sun SY. Role of acetylcholine, corticoids and opioids in the rostral ventrolateral medulla in stress-induced hypertensive rats. Biol Signals 1995;4:124-32.
- Lin Q, Li P. Rostral medullary cholinergic mechanisms and chronic stress-induced hypertension. J Auton Nerv Syst 1990;31:211-8.
- Hwang IS, Hwang WC, Wu JN. Effect of fructose-induced hypertension on the renin-angiotensin-aldosterone system and atrial natriuretic factor. Am J Hypertens 1989;2:424-7.
- Iyer SN, Raizada MK, Katovich MJ. AT1 receptor density changes during development of hypertension in hyperinsulinemic rats. Clin Exper Hypertens 1996;18:793-810.
- Wang DH, Wu W, Lookingland KJ. Degeneration of capsaicin-sensitive sensory nerves leads to increased salt sensitivity through enhancement of sympathoexcitatory response. Hypertension 2001;37:440-3.
- Huang Y, Wang DH. Role of AT1 and AT2 receptor subtypes in salt-sensitive hypertension induced by sensory nerve degeneration. J Hypertens 2001;19:1841-6.
- Fregly MJ, Kikta DC, Threatte RM, Torres JL, Barney CC. Development of hypertension in rats during chronic exposure to cold. J Appl Physiol 1989;66:741-9.
- Sun Z, Fregly MJ, Cade R. Effect of renal denervation on elevation of blood pressure in cold-exposed rats. Can J Physiol Pharmacol 1995;73:72-8.
- Sun Z, Cade R, Tatum C. Central imidazoline and angiotensin II receptors in cardiovascular responses to chronic cold exposure in rats. J Therm Biol 2001;26:513-8.
- Sun Z, Cade R, Morales C. Role of central angiotensin II receptors in cold-induced hypertension. Am J Hypertens 2002;15:85-92.
- Sun Z, Cade R, Katovich MJ, Fregly MJ. Body fluid distribution in rats with cold-induced hypertension. Physiol Behav 1999;65:879-84.
- Sun Z, Cade R. Cold-induced hypertension and diuresis. J Thermal Biol 2000;25:105-9.
- Thorvaldsen P, Asplund K, Kuulasmaa K, Rajakangas AM, Schroll M. Stroke incidence, case fatality, and mortality in the WHO MONICA project: World Health Organization Monitoring Trends and Determinants in Cardiovascular Diseases. Stroke 1995;26:361-7.
- Donaldson GC, Robinson D, Allaway SL. An analysis of arterial disease mortality and BUPA health screening data in men, in relation to outdoor temperature. Clin Sci Colch 1997;92:261-8.
- Gyllerup S, Lanke J, Lindholm LH, Schersten B. Cold climate is an important factor in explaining regional differences in coronary mortality even if serum cholesterol and other established risk factors are taken into account. Scott Med J 1993;38:169-72.
- Sheth T, Nair C, Muller J, Yusuf S. Increased winter mortality from acute myocardial infarction and stroke: the effect of age. J Am Coll Cardiol 1999;33:1916-9.
- He BL. Epidemiological characteristic of stroke in 1985–1989, Beijing. Chin J Med 1993;73:104-8.
- Marchant B, Ranjadayalan K, Stevenson R, Wilkinson P, Timmis AD. Circadian and seasonal factors in the pathogenesis of acute myocardial infarction: the influence of environmental temper-ature. Br Heart J 1993;69:385-7.
- Papanek PE, Wood CE, Fregly MJ. Role of the sympathetic nervous system in cold-induced hypertension in rats. J Appl Physiol 1991;71:300-6.
- Flaim SF, Hsieh C. Effect of cold acclimatization on rabbit carotid artery: altered response to norepinephrine. Gen Pharmacol 1978;9:437-42.
- Fregly MJ, Brummermann M. Effect of chronic exposure to cold on vascular responsiveness to phenylephrine and angiotensin II. Pharmacology 1993;47:237-43.
- Shechtman O, Fregly MJ, van Bergen P, Papanek PE. Prevention of cold-induced increase in blood pressure of rats by captopril. Hypertension 1991;17:763-70.
- Peng JF, Kimura B, Fregly MJ, Phillips MI. Reduction of cold-induced hypertension by antisense oligodeoxynucleotides to angiotensinogen mRNA and AT1-receptor mRNA in brain and blood. Hypertension 1998;31:1317-23.
- Sun Z, Cade R, Zhang Z, Alouidor J, Van H. Angiotensinogen gene knockout delays and attenuates cold-induced hypertension. Hypertension 2003;41:322-7.
- van Bergen P, Fregly MJ, Papanek PE. Effect of a reduction in sodium intake on cold-induced elevation of blood pressure in the rat. Proc Soc Exp Biol Med 1992;200:472-9.
- Sellye H. Production of nephrosclerosis by overdosage with deoxycorticosterone acetate. Can Med Assoc J 1942;47:515-9.
- Crofton JT, Share L, Shade RE. The importance of vasopressin in the development and maintenance of DOC-salt hypertension in the rat. Hypertension 1978;1:31-8.
- Intengan HD, Park JB, Schiffrin EL. Blood pressure and small arteries in DOCA-salt-treated genetically AVP-deficient rats. Role of endothelin. Hypertension 1999; 34 [part2]: 907–13.
- Mohring J, Mohring B, Petri M, Haack D. Vasopressor role of ADH in the pathogenesis of malignant DOC hypertension. Am J Physiol 1977;232:F260-9.
- Katholi RE, Naftilan AJ, Oparil S. Importance of renal sympathetic tone in the development of DOCA-salt hypertension in the rat. Hypertension 1980;2:266-73.
- O’Hagan KP, Thomas GD, Zambraski EJ. Renal denervation decreases blood pressure in DOCA-treated miniature swine with established hypertension. Am J Hypertens 1990;3:62-4.
- Wilson KM, Sumners C, Hathaway S, Fregly MJ. Mineralocorticoids modulate central angiotensin II receptors in rats. Brain Res 1986;382:87-96.
- French JF, Anderson BA, Downs TR, Dage RC. Dual inhibition of angiotensin-converting enzyme and neutral endopeptidase in rats with hypertension. J Cardiovasc Pharmacol 1995;26:107-13.
- Wong PC, Price WA Jr, Chiu AT, Duncia JV, Carini DJ, Wexler RR. In vivo pharmacology of DuP 753. Am J Hypertens 1991;4:S288-98.
- Van den Berg DT, de Kloet ER, de Jong W. Central effect of mineralocorticoid antagonist RU-28318 on blood pressure of DOCA-salt hypertensive rats. Am J Physiol 1994;267:E927-33.
- Matsumura Y, Hashimoto N, Taira S, Kuro T, Kitano R, Ohkita M, et al. Different contribution of endothelin-A and endothelin-B receptors in the pathogenesis of deoxycorticosterone acetate-salt-induced hypertension in rats. Hypertension 1999;33:759-65.
- Moreau P, Schifrin EL. Role of endothelins in animal models of hypertension: focus on cardiovascular protection. Can J Physiol Pharmacol 2003;81:511-21.
- Schiffrin EL. Role of endothelin-1 in hypertension and vascular disease. Am J Hypertens 2001;14:S83-9.
- van den Meiracker AH. Endothelins and venous tone in DOCA-salt hypertension. J Hypertens 2002;20:587-9.
- Callera GE, Montezano AC, Touyz RM, Zorn TMT, Carvalho MHC, Fortes ZB, et al. ETA receptor mediates altered leukocytes-endothelial cell interaction and adhesion molecules expression in DOCA-salt rats. Hypertension 2004;43:872-9.
- Li L, Chu Y, Fink GD, Engelhardt JF, Heistad DD, Chen A. Endothelin-1 stimulates arterial VCAM-1 expression via NADPH oxidase-derived superoxide in mineralocorticoid hypertension. Hypertension 2003;42:997-1003.
- Manning RD Jr, Meng S, Tian N. Renal and vascular oxidative stress and salt-sensitivity of arterial pressure. Acta Physiol Scand 2003;179:243-50.
- Goldblatt H, Lynch J, Hanzel RF. Studies on experimental hypertension. The production of persistent elevation of systolic blood pressure by means of renal ischemia. J Exp Med 1934;59:347-79.
- Zandberg P. Animal models in experimental hypertension: relevance to drug testing and discovery. In: van Zwieten PA, editor. Handbook of hypertension. Vol. 3: Pharmacology of antihypertensive drugs. New York: Elsevier Science Publishers; 1984. Chapter 2.
- Leenen FHH, De Jong W. Plasma renin and sodium balance during development of moderate and severe renal hypertension in rats. Circ Res 1975;36/37 Suppl 1:179-84.
- Liard JF, Cowley AW Jr, McCaa RE, McCaa CS, Guyton AC. Renin, aldosterone body fluid volumes and the baroreceptor reflex in the development and reversal of Goldblatt hypertension in conscious dogs. Circ Res 1974;34:549-60.
- Sawamura T, Nakada T. Role of dopamine in the striatum, renin-angiotensin system and renal sympathetic nerve on the development of two-kidney, one clip Goldblatt hypertension. J Urol 1996;155:1108-11.
- Bean BL, Brown JJ, Casals-Stenzel J, Fraser R, Lever AF, Millar JA, et al. The relation of arterial pressure and plasma angiotensin II concentration. A change produced by prolonged infusion of angiotensin II in conscious dog. Circ Res 1979;44:452-8.
- Freeman RH, Davis JO, Watkins BE. Effects of continuous converting enzyme blockade on renovascular hypertension in the rat. Am J Physiol 1979;236:F221-4.
- Brown JJ, Fraser R, Lever AF. Mechanism in hypertension: a personal view. In: Genest J, Koiw E, Kuchel O, editors. Hypertension. New York: McGraw-Hill; 1977. Chapter 12.1.
- Ten Berg RGM, Leenen FHH, De Jong W. Plasma renin activity and sodium, potassium and water excretion during reversal of hypertension in the one-clip, two kidney hypertensive rat. Clin Sci 1979;57:7-15.