
Functional role of anion channels in cardiac diseases1
Introduction
Abnormalities of cardiac ion channels have been linked to a variety of inherited and acquired cardiac diseases including myocardial ischemia, hypertrophy, heart failure, and arrhythmias[1–5]. In addition, ion channels may also be mediators of the cardioprotective effects of ischemic preconditioning (IPC)[6,7]. While cation (K+, Na+, and Ca2+) channels have received the most attention in the past four decades, the role of anion channels in the cardiovascular system has been largely ignored. Within the last 15 years, a re-surgence of interest in Cl– channels in the cardiovascular system has led to the discovery of at least seven different types of Cl– currents in cardiac cells from different regions of the heart and in different species[8]. Intensive efforts have been given to characterize the properties of these anion channels at the cellular and molecular levels. More details about the biophysical, pharmacological, and molecular properties of Cl– channels in the heart can be found in several recent excellent review articles[8–11]. It has also been demonstrated in recent studies that Cl– channels may be involved in the regulation of a large repertoire of cellular functions, including cellular excitability, cell volume homeostasis, intracellular organelles acidification, cell migration, proliferation and differentiation, and apoptosis[8,9,12]. With the recent identification of molecular entities responsible for cardiac Cl– channels[8] and the genes mapped to specific human chromosomal locations[13], gene targeting and transgenic techniques have been used to delineate the functional role of Cl– channels in the context of health and disease. It has been reported that Cl– channels could contribute to: 1) arrhythmogenesis in myocardial injury; 2) the adaptive remodeling of the heart during myocardial hypertrophy and heart failure; and 3) IPC. Therefore, anion channels represent very attractive novel targets for therapeutic approaches to the treatment of heart diseases. In this review, we will briefly summarize the major findings and recent advances in the study of functional role of anion channels in the heart.
Anion channels in the heart
Since the independent discovery of a cAMP-activated Cl– current in the guinea pig heart by Bainski et al, Harvey and Hume in 1989[14,15], intensive efforts have been made to characterize Cl– channels in the cardiovascular system at both the cellular and molecular levels. These have been recently reviewed and thoroughly described elsewhere[8–11] and will not be repeated in this review. Briefly, at the molecular level, all cardiac Cl– channels described so far may fall into the following Cl– channel gene families (Figure 1): 1) the cystic fibrosis transmembrane conductance regulator (CFTR), which is a member of the adenosine triphosphate-binding cassette (ABC) superfamily and may be responsible for the Cl– currents activated by protein kinase A (PKA) (ICl.PKA)[14–16], protein kinase C (PKC) (ICl.PKC)[17,18], and extracellular ATP (ICl.ATP) in the heart[19–21]; 2) ClC voltage-gated Cl– channel superfamily: a) ClC-2, which is responsible for the hyperpolarization- and cell swelling-activated inwardly rectifying Cl-–current (ICl.ir)[22–24]; b) ClC-3, which is responsible for the volume regulated outwardly rectifying Cl– current (ICl.vol), including the basally-activated (ICl.b)[25] and swelling-activated (ICl.swell) components[25–34]; 3) CLCA-1, which was thought to be responsible for the Ca2+-activated Cl– current (ICl.Ca)[35–38]; and 4) Bestrophin, a new candidate for ICl.Ca[39–42]. Further studies on the molecular and functional properties of these Cl– channel genes are necessary to define the structure of the channel proteins and to elucidate the physiological and clinical significance of these channels.
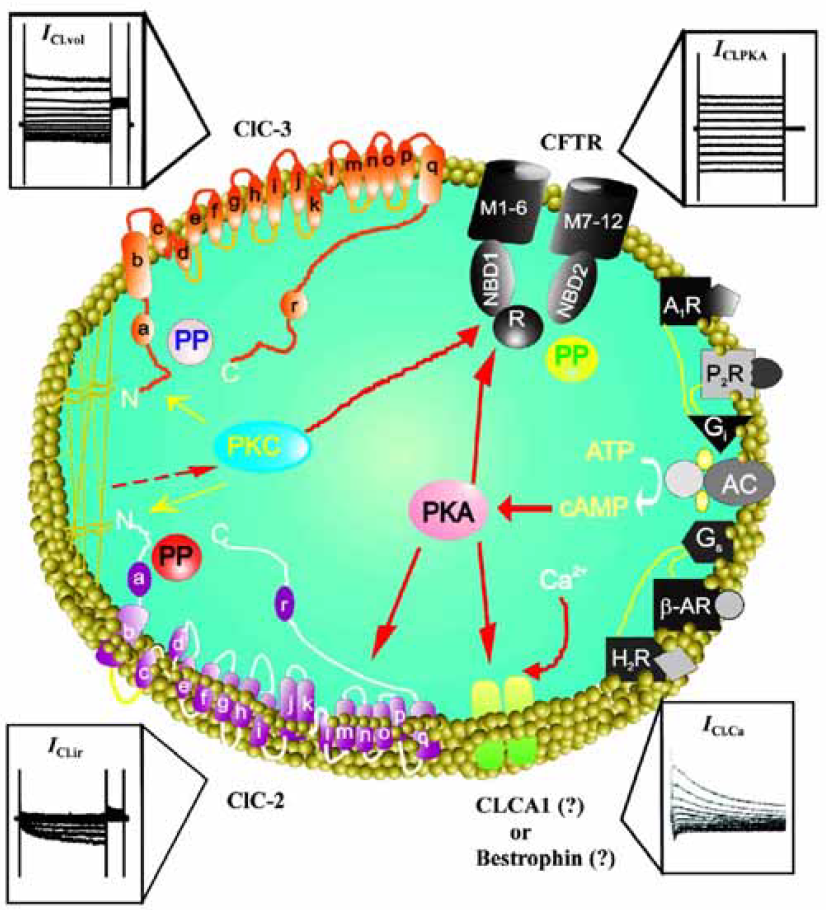
Functional role of Cl– channels in cardiac diseases
Theoretically, Cl– channels could be involved in the regulation of cellular excitability, cell volume homeostasis, intracellular organelles acidification, cell proliferation and differentiation, and apoptosis[12]. Thus, they may have important physiological and pathological significance in cardiac function under normal and pathological (hypoxia, ischemia, myocardial infarction, hypertrophy, and heart failure) conditions. Mutations in several Cl– channels have been known to result in human inherited diseases[13]. But the exact role of Cl– channels in human cardiovascular physiology and pathophysiology is still unclear[8]. The ability to examine the exact role of Cl– channels in human cardiovascular physiology and pathology has been hampered by the lack of specific pharmacological and molecular tools. With the recent identification of the molecular entities responsible for Cl– channels in the heart[8] and the genes mapped to specific human chromosomal locations[13], it is now possible to overcome these obstacles by use of gene targeting and trans-genic animals. We have been using a multitude of approaches from traditional methodologies including bio-physics, biochemistry, electrophysiology, and pharmacology to state-of-the-art technologies including telemetry system, echocardiography, genomics, and proteomics to effectively and accurately define the role of each Cl– channel in heart function in the context of health and disease.
Functional role in electrophysiology and arrhythmogenesis
Estimates of intracellular Cl– activity (aiCl) in cardiac myocytes from ion-selective microelectrode studies indicate the equilibrium potential for Cl– (ECl) be more positive than the resting membrane potential under normal physiological conditions with an extracellular Cl– concentration ([Cl–]o) of 145 mmol/L and an intracellular Cl– concentration ([Cl–]i) of 10 to 20 mmol/L[43–46]. Because the ECl is within a membrane potential range (usually –65 to –40 mV) that can be either negative or positive to the actual membrane potential during the normal cardiac cycle, activation of cardiac Cl– channels can generate both inward and outward currents (Figure 2). Thus, compared with cationic channels, Cl– channels have the unique ability to cause both depolarization as well as repolarization during the action potential and produce significant effects on cardiac pacemaker activity and action potential characteristics.
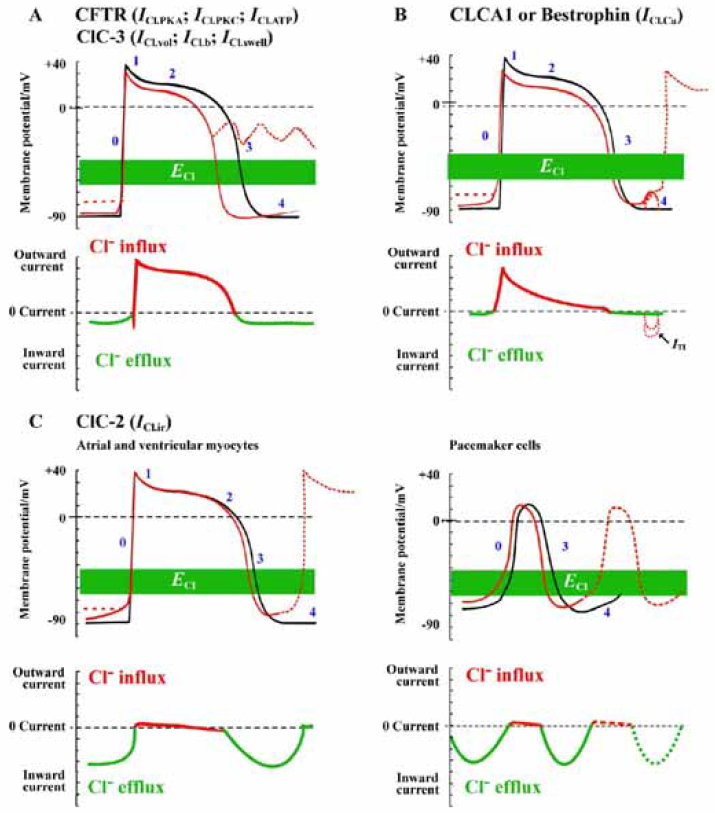
The degree to which activation of Cl– currents depolarizes the resting membrane or accelerates the repolarization of action potential depends critically on the actual value of ECl and the magnitude of the Cl– conductance relative to the total membrane conductance. Under physiological conditions the transmembrane Cl– gradient is asymmetrical. Thus, the activation of CFTR and ClC-3 Cl– channels in the heart will result in outwardly rectifying currents. This will have more significant effects at positive potentials to accelerate repolarization and shortening of the action potential duration compared with smaller depolarizing effects at negative potentials near the resting membrane potential (Figure 2). The ability of Cl– current activation to depolarize cardiac cells is also opposed by the presence of a large background K+ conductance that normally controls the resting membrane potential. Both abbreviation of APD and depolarization of Em upon activation of Cl– channels may play a role in rhythm disturbance and likely contribute to arrhythmogenesis under pathological conditions.
CFTR and arrythmogenesis CFTR channels are closed under basal conditions but can be open under conditions where intracellular PKA- and PKC-dependent phosphorylation activity is increased. A major physiological role of activation of CFTR channels may be to minimize (oppose) the significant action potential prolongation associated with β-adrenergic stimulation of ICa. This is expected to contribute to action potential shortening during strong adrenergic stimulation and faster heart rates. Therefore, activation of CFTR channels may prevent excessive prolongation of APD and protect the heart against the development of early after depolarizations (EAD) and triggered activity caused by activation of Ca2+ channels in the presence of β-adrenergic stimulation. EAD arising from phase 2 and 3 underlie focal triggered tachyarrhythmias and repolarization abnormalities, which contribute to cardiac sudden death[47]. It is well-established that APD prolongation favors EAD by allowing recovery of inward currents and, conversely, shortening of APD makes it more difficult to induce EAD. Therefore, activation of CFTR channels should protect against focal triggered arrhythmias. However, when background K+ conductance is reduced in the case of myocardial hypokalemia, activation of CFTR channels will cause significant membrane depolarization and induce abnormal automaticity leading to the development of, EAD (dotted red lines in Figure 2A). These predicted effects of CFTR channel activation on APD and automaticity have been verified experimentally by manipulations of the Cl– gradient or the use of Cl– channel blockers[11,48–51]. Histamine was found to activate CFTR channels in ventricular myocytes and induce oscillatory activity and abnormal impulses in the heart, although the contribution of CFTR channels to these arrhythmogenic activities has not been further explored. It has been shown that activation of CFTR channels contributes to hypoxia-induced shortening in APD[52]. Activation of CFTR channels may accelerate the development of reentry because of the shortening of APD and refractoriness and a decrease in conduction velocity caused by a slight depolarization of diastolic potential leading to Na+ channel inactivation.
ClC-3 and arrhythmogenesis The current through ClC-3 channels under basal or isotonic conditions is small, but can be further activated by stretching of the cell membrane by inflation and/or cell swelling induced by exposure to hypoosmotic solutions. Activation of ClC-3 channels is expected to induce a similar effect on cardiac action potentials as that of activation of CFTR channels (Figure 2A) because both Cl– currents through both channels are relatively time- and voltage-independent over the physiological range of membrane potentials[53,54]. Activation of ClC-3 channels might produce more significant action potential shortening than CFTR channels because of its stronger outwardly rectifying property. Because myocardial cells swell during hypoxia and ischemia, and the washout of hyperosmotic extracellular fluid after reperfusion induces further cell swelling, activation of ClC-3 channels may also contribute to hypoxia, ischemia and reperfusion induced shortening in APD and arrhythmias[9,53,54]. Abbreviation of APD and, therefore, the effective refractory period reduces the length of the conducting pathway needed to sustain reentry (wavelength). In principle, this favors the development of atrial or ventricular fibrillation, which depends on the presence of multiple reentrant circuits or rotating spiral waves. ICl.swell also may slow or enhance the conduction of early extrasystoles, depending on the timing. In the case of myocardial hypertrophy and heart failure, ionic remodeling is one of the major features of pathophysiological changes[55]. It has been found that the current densities and molecular expression of several major repolarizing K+ channels (such as Kv4x) are significantly reduced, which may be responsible for the prolongation of APD and development of EAD[55]. However, under these conditions, ICl.vol is constitutively active[56]. The persistent activation of ICl.vol may limit the APD prolongation and make it more difficult to elicit EAD. Indeed, as shown in Figure 3, in myocytes from hearts in failure, block of ICl.vol by tamoxifen significantly prolonged APD and decreased the depolarizing current required to elicit EAD by about 50% (Figure 4B) and hyperosmotic cell shrinkage, which also inhibits ICl.vol, was almost equivalent to tamoxifen in causing EAD in these myocytes (Figure 4C)[9]. Therefore, the consequences of activation of ICl.vol are very complex. It may be detrimental, beneficial, or simultaneously both in different parts of the heart.
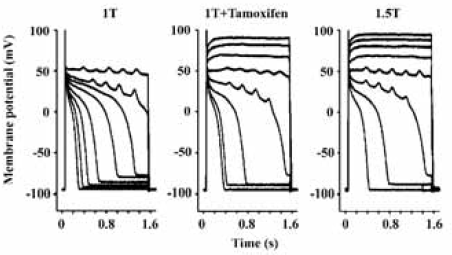
It has been shown that mechanical stretching or dilation of the atrial myocardium is able to cause arrhythmias. Since ICl.swell was also found in sino-atrial (S-A) nodal cells, ClC-3 channels may serve as a mediator of mechanotransduction and play a significant role in the pacemaker function if they act as the stretch activated channels in these cells[9,57]. Baumgarten’s laboratory has recently demonstrated that ICl.swell in ventricular myocytes could be directly activated by mechanical stretch through selectively stretching β1-integrins with mAb-coated magnetic bEAD[9,58,59]. Although it has been suggested that stretch and swelling activate the same anion channel in some non-cardiac cells, further study is needed to determine whether this is true in cardiac myocytes.
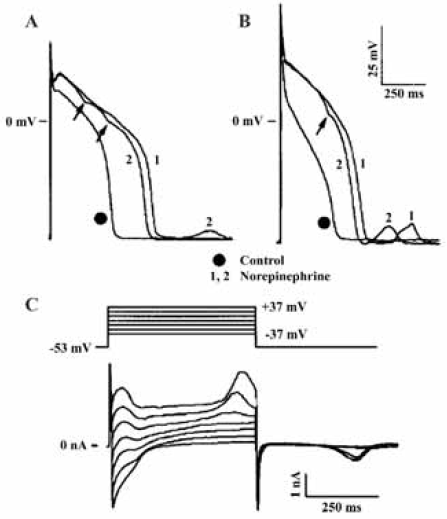
Ca2+-activated Cl– channel and arrhythmogenesis As illustrated in Figure 2B, the activation of ICl.Ca will have considerably different effects on cardiac action potentials and resting membrane potential from those of CFTR and ClC-3 channels, even though ICl.Ca is also expected to be outwardly rectifying under physiological conditions. This is because the kinetic behavior of ICl.Ca is significantly determined by the time course of the [Ca2+]i transient[60]. Normally, ICl.Ca will have insignificant effects on the diastolic membrane potential, as resting [Ca2+]i is low. When [Ca2+]i is substantially increased above the physiological resting level, however, ICl.Ca carries a significant amount of transient outward current. ICl.Ca will be activated early during the action potential in response to an increase in [Ca2+]i associated with Ca2+-induced Ca2+ release (CICR). The time course of decline of the [Ca2+]i transient will determine the extent to which ICl.Ca contributes to early repolarization during phase 1 (Figure 2B). In the rabbit left ventricle, ICl.Ca contributes to APD shortening in subendocardial myocytes but not in subepicardial myocytes. These differences in functional expression of ICl.Ca may reduce the electrical heterogeneity in the left ventricle[61]. In Ca2+-overloaded cardiac preparations, ICl.Ca can contribute to the arrhythmogenic transient inward current (ITI, Figure 2B)[62]. ITI produces delayed after-depolarization (DAD)[63] and induces triggered activity (red dotted line in Figure 2B), which is an important mechanism for abnormal impulse formation. In sheep Purkinje and ventricular myocytes, activation of ICl.Ca was found to induce DAD and plateau transient repolarization (Figure 4)[64]. Therefore, blockade of ICl.Ca may be potentially antiarrhythmogenic by reducing DAD amplitude and triggered activity based on DAD. However, the role of ICl.Ca in phase 1 repolarization and the generation of EAD and DAD of either normal or failing human heart seem very limited[65–67]. Therefore, the clinical relevance of ICl.Ca blockers remains to be determined.
ClC-2 and arrhythmogenesis ClC-2 channels are activated by hyperpolarization, cell swelling, and acidosis and have an inwardly rectifying I-V relationship. During the cardiac action potential, therefore, the ClC-2 channel will conduct a mainly inward current as a result of Cl– efflux at negative membrane potentials and cause a depolarization of the resting membrane potential of cardiac cells. At membrane potentials more positive than ECl, ClC-2 may conduct a small outward current as a result of Cl- influx and may accelerate repolarization of the action potential. It is also possible that, in a manner analogous to the role and tissue distribution pattern of the cationic pacemaker channels (If), Cl.ir channels normally play a much more prominent role in the SA or AV nodal regions of the heart (Figure 2C). The hyperpolarization-activated inward rectifying Cl– current (ICl.ir) through ClC-2 channels under basal or isotonic conditions is small, but can be further activated by hypotonic cell swelling[22] and acidosis[23,24]. The volume-sensitivity of the channel also suggests its role in cell volume regulation. The sensitivity of ClC-2 to [H+]o and cell volume may be of pathological importance during hypoxia- or ischemia-induced acidosis or cell swelling. Therefore, it may be possible that the significance of ICl.ir in the heart becomes more prominent under some pathological conditions (ischemia or hypo-xia)[68]. As a matter of fact, ischemia and acidosis have consistently been shown to depolarize the resting membrane potential of cardiac myocytes, increase automaticity and cause lethal arrhythmias, although the mechanism has remained obscure[1,11]. It is reasonable to suggest that an increase in ClC-2 conductance could be responsible for these phenomena and be pro-arrhythmic. Drugs targeting ClC-2 channels could be anti-arrhythmic. Therefore, the ClC-2 channels could have important clinical significance for such cardiac diseases as arrhythmias, ischemia and reperfusion, and congestive heart failure. Activation of ClC-2 current should mainly cause a depolarization of the RMP and it is suggested that the acidosis-induced increase in ICl,ir might underlay the depolarization of the resting membrane potential during acidosis or hypoxia[23,24].
It should be pointed out that prediction of the consequences of activation of Cl– channels is complex. Most studies that have examined the contribution of Cl– currents to the cardiac action potential and arrhythmias have relied on anion antagonist and substitution experiments. The pharmacological specificity of many of these anion channel antagonists can be problematic, and anion substitution, in addition to altering anion movement through channels, can have other unpredictable side effects on other transport proteins and signaling pathways[69,70]. With the recent identification of the molecular entities responsible for Cl– channels in the heart, it is now possible to combine electrophysiological, molecular biological, and especially gene-targeting techniques in the study of cardiac Cl– channels to effectively and accurately define the role of each Cl– channel in heart function. However, as the distribution of various Cl– channels in the heart varies among cell types and regions[8], activation of these channels may increase the dispersion of the electrophysiological properties and provide substrates for heart diseases involving cardiac arrhythmias and myocardial remodeling.
Functional role of Cl– channels in cardiac IPC
Ischemia causes myocardial damage and leads to infarction through apoptosis (programmed cell death) and necrosis. IPC is a phenomenon in which brief ischemic episodes elicit protection of the heart against sustained ischemia. It has been suggested that both sarcolemmal and mitochondrial ATP-sensitive potassium channels (sarc-KATP and mito-KATP, respectively) may serve as triggers or end-effectors. PKC may link cellular signal events during ischemia to the activation of end-effectors, which will somehow prevent or delay apoptosis and protect the cardiac myocytes. The precise mechanism of IPC, however, remains to be elucidated. Several recent studies have pointed to a potential role of Cl– channels in IPC.
ICl.swell and ClC-3 in IPC It has been reported that the block of ICl.swell in rabbit cardiac myocytes inhibits preconditioning by brief ischemia, hypoosmotic stress[71,72] and adenosine receptor agonists[73]. These studies are solely based on the use of several Cl– channel blockers, such as anthracene-9-carboxylic acid (9-AC) and 4-acetamide-4'-isothiocyanato-stilbene-2,2'-disulfonic acid (SITS). As mentioned above, these pharmacological tools lack specificity to a particular Cl– channel in the heart and may also act on other ion channels or transporters. Therefore it has been very difficult to confirm the causal role of ICl.swell in IPC[74]. The exact role of ICl.swell in IPC needs to be further determined adequately using more specific approaches. To specifically test whether the volume-regulated Cl– channels are indeed involved in IPC, we have recently established in vitro and in vivo IPC models in ClC-3 knockout mice (ClCn3–/–). Our preliminary results indicate that targeted inactivation of ClC-3 gene prevented protective effects of late IPC but not of early IPC, suggesting that ICl.swell may contribute differently to early and late IPC[75]. The underlying mechanisms for these differential effects are currently unknown. Recent reports, however, suggest that ICl.swell and ClC-3 might play an important role in apoptosis. Cl– channel blockers 4,4'-diisothio-cyano-stilbene-2,2'-disulphonate (DIDS) and 5-nitro-2-(3-phenylpropyllamino)-benzoic acid (NPPB) were as potent as a broad-spectrum caspase inhibitor in preventing apoptosis and elevation of caspase-3 activity and improved cardiac contractile function after ischemia and in vivo reperfu-sion[76]. Transgenic mice overexpressing Bcl-2 in the heart had significantly smaller infarct size and reduced apoptosis of myocytes after ischemia and reperfusion[77]. It has been shown that Bcl-2 induces up-regulation of ICl.vol by enhancing ClC-3 expression in human prostate cancer epithelial cells[78]. Cell shrinkage is an integral part of apoptosis, suggesting that ICl.vol and ClC-3 might be intimately linked to apoptotic events through regulation of cell volume homeostasis[78,79].
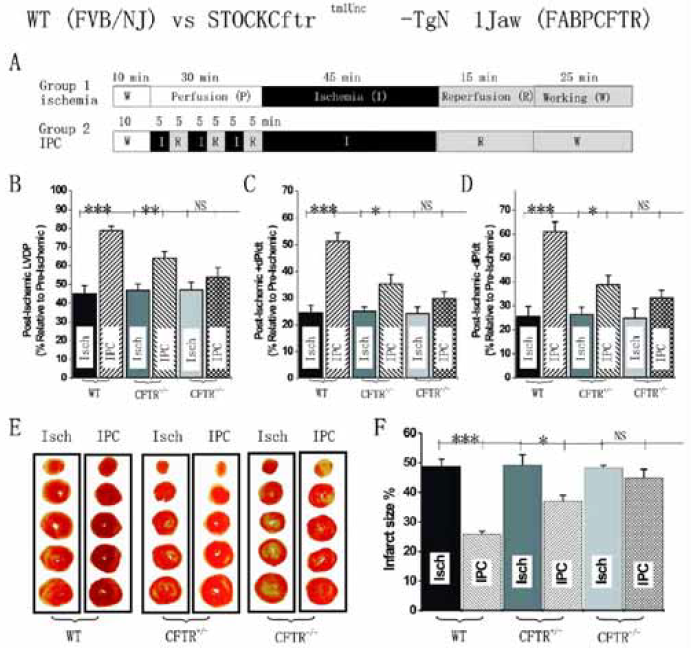
CFTR channels and IPC Several lines of evidence suggest that CFTR channels could be involved in IPC including: (1) sarc-KATP blockers, such as glibenclamide, which suppress IPC protection, also block CFTR channels in noncardiac[80,81] and cardiac cells[19,82]; (2) PKC and PKA, two essential second messengers in IPC[83,84] can activate CFTR channels[8,19,85]; and (3) triggers of IPC (nitric oxide, opioids, and adenosine etc) can all regulate CFTR channel function[8]. We have directly tested whether activation of CFTR channels is involved in IPC by studying hemodynamics and tissue injury of hearts isolated from WT and two strains of CFTR knockout (CFTR–/–) mice subjected to ischemia and reperfusion. In isolated mouse heart perfused in the Langendorff or working heart mode, we have recently found that targeted inactivation of CFTR gene prevented protection on cardiac function and myocardium injury against sustained ischemia by ischemic preconditioning (Figure 5)[86]. Our in vivo studies using both wild type and CFTR knockout mice also demonstrated that CFTR was an important mediator in both early and late ischemic preconditioning in the heart[87]. Several mechanisms may be responsible for a functional role of CFTR channels in mouse heart IPC: (1) It has been demonstrated that cardiac CFTR plays a role in early action potential shortening during hypoxia and ischemia[52]. Activation of CFTR will also decrease resting membrane potential and action potential duration, thereby limiting intracellular Ca2+ overload and cell damage[8]; (2) The CFTR channel is an important transporter of sphingosine 1-phosphate (S-1-P)[88], which has recently emerged as an important lipid messenger involved in IPC[89]; (3) CFTR is permeable not only to Cl–, but also to larger organic ions, as well as reduced and oxidized forms of glutathione (GSH)[90]. Therefore CFTR may contribute to the control of oxygen stress-induced apoptosis and the regulation of inflammation and the immune responses; (4) CFTR might decrease intracellular pH and modulate apoptosis[91]; (5) CFTR functions as a regulator of volume-dependent homeostatic cell mechanisms in cell proliferation and apoptosis[92]. We are currently in the process of investigating these potential mechanisms and the relative role of CFTR in early and late preconditioning.
ICl.Ca in IPC It has been well known that ischemia/reperfusion usually causes a cytosolic overload of Ca2+ in cardiac myocytes[93,94]. Therefore, it is very possible that ICl.Ca may be activated during ischemia and reperfu-sion[38,62,64,95–98]. But, no information for the possible involvement of ICl.Ca in IPC is currently available.
Functional role of Cl– channels in myocardial hypertrophy and heart failure Myocardial hypertrophy and its progression to dilated cardiomyopathy or heart failure are characterized by not only structural remodeling, including hypertrophic growth of cardiac myocytes (changes in cell volume) and changes in the cytoskeleton and extracellular matrix (ECM)[99,100] but also ionic remodeling, that is, changes in expression and activity of many ion channels. It should be pointed out that ionic remodeling during the progression of hypertrophy to heart failure provides not only substrates for arrhythmias but also cellular mechanisms for structural remodeling. During the remodeling process, multiple neurohormonal and intracellular signaling cascades, including tyrosine kinases, PKA, PKC, protein phosphatases, MAP kinases, and endothelin, are activated[101]. These second messengers are well-known effective regulators of various ion channels. Indeed, it has been found that several cation channels, such as K+ channels, Ca2+ channels, and stretch-activated non-selective channels, undergo significant changes. Recent evidence also supports possible involvement of anion channels in the remodeling process.
ICl.swell and ClC-3 in myocardial hypertrophy and heart failure ICl.swell is persistently activated in ventricular myocytes from a canine pacing-induced dilated cardiomyopathy model[102]. Using the perforated patch-clamp technique, Clemo et al found that, even in isotonic solutions, a large 9-AC-sensitive, outwardly rectifying Cl– current was recorded in heart failure myocytes but not in normal myocytes. Graded hypotonic cell swelling (90%−60% hypotonic) failed to activate additional current while graded hypertonic cell shrinkage caused an inhibition of the “basal” Cl– current in failure myocytes. Moreover, the maximum current density of the ICl.swell in failure myocytes was about 40% greater than that in osmotically swollen normal myocytes. Constitutive activation of ICl.swell is also observed in several other animal models of heart failure, such as a rabbit aortic regurgitation model of dilated cardiomyopathy[103,104] and a dog model of heart failure caused by myocardial infarction[105]. In human atrial myocytes obtained from patients with right atrial enlargement and/or elevated left ventricular end-diastolic pressure, a tamoxifen sensitive ICl.swell was also found to be persistently activated[106]. It is not known at this time whether ICl.swell is also persistently activated in hypertrophied non-failure (or non-dilated) myocytes in the above described models or in the human heart. In a rat aortic constriction model, however, a 9-AC-sensitive Cl– current is present in hypertrophied ventricular myocytes but not in control myocytes, and this hypertrophy-activated Cl– current seems to contribute to the shortening of APD in the hypertrophied cells[107]. It is not known, however, whether this hypertrophy-activated Cl– current is the same as ICl.swell because the volume-sensitivity of this Cl– current was not assessed. Nevertheless, it is possible that persistent activation of ICl.swell is a common response of cardiac myocytes to hypertrophy or heart failure-induced remodeling. The mechanism for this phenomenon is still not clear. Perhaps the cell volume increase caused by hypertrophy and cell membrane stretch caused by dilation, are both involved in the activation of ICl.swell. Alternatively, the persistent activation of ICl.swell may be caused by signaling cascades activated during hypertrophy and heart failure independent of changes in cell length and volume, or both. ICl,swell could be activated by direct stretching of β1-integrin through focal adhesion kinase (FAK) and/or Src[58]. Mechanical stretch of myocytes also releases angiotensin II (AngII), which binds to AT1 receptors (AT1R) and stimulates FAK and Src in an autocrine-paracrine loop. A recent study by Browe and Baumgarten suggests that the stretch of β1-integrin in cardiac myocytes activates ICl.swell by activating AT1R and NADPH oxidase and, thereby, producing reactive oxygen species. In addition, NADPH oxidase may be intimately coupled to the channel responsible for ICl.swell, providing a second regulatory pathway for this channel through membrane stretch or oxidative stress[59]. This finding is very important for further understanding of the mechanism for hypertrophy activation of ICl.swell and ClC-3 channels and their relationship to hypertrophy and heart failure as it is very well known that Ang II plays a crucial role in myocardial hypertrophy and heart failure[108].
The functional and clinical significance of ICl.swell in the hypertrophied and dilated heart is currently unknown. Using a mouse aortic banding model of myocardial hypertrophy, we have recently found that targeted disruption of ClC-3 gene (ClCn3–/–) accelerated the development of myocardial hypertrophy and the discompensatory process[109], suggesting that activation of ICl.vol might be important in the adaptive remodeling of the heart during pressure overload. Further studies on the mechanism for the ClC-3 channels’ effects on hypertrophy and heart failure are in progress in our laboratory. It is well accepted that in most cells activation of ICl.vol represents one important trigger to initiate regulatory volume decrease (RVD) when cells swell[12]. Cell volume homeostasis, therefore, could be an important function of ICl.swell activation in the heart. Activation of Cl– conductance causes significant changes in APD and intracellular Ca2+ concentration, and should also affect excitation-contraction (E-C) coupling, contractility, and other hemodynamic functions of the heart[8,11]. Recent studies suggest that ICl.swell and ClC-3 channels play important roles in cell prolifera-tion[110], differentiation[111], migration[112], and apoptosis[78,79]. All of these have been demonstrated as important cellular processes in myocardial remodeling during hypertrophy and heart failure[113].
CFTR in myocardial hypertrophy and heart failure Remodeling of CFTR channels has been observed in myocardial hypertrophy and heart failure. Using in situ mRNA hybridization in a combined pressure and volume overload model of heart failure in the rabbit, Wong et al found that the normal epicardial to endocardial gradient of CFTR mRNA expression is reversed due to a significant decrease in epicardial expression of CFTR mRNA in the rabbit left ventricle[114]. A post-translational change in the CFTR expression could be responsible for this phenomenon[115]. The loss of the normal transmural gradient of repolarising ion channels is likely to contribute to instability of repolarisation in the hypertrophied heart and hence increased risk of cardiac arrhythmias in patients with heart failure. The exact functional and clinical significance of the changes in CFTR expression during hypertrophy and heart failure is currently not clear and merits further study.
ICl.Ca in myocardial hypertrophy and heart failure The critical role of Ca2+ in cardiac development, function, and disease is undisputable. Despite the heterogeneous etiology and overt manifestations of heart failure, abnormalities in Ca2+ handling are prominent, and alterations in Ca2+ homeostasis are a hallmark of myocardial hypertrophy and heart failure[116]. Ca2+ transients in failing cardiac myocytes, for example, are characterized by diminished amplitude, elevated diastolic Ca2+ levels, and prolonged decay of the Ca2+ transients. In non-cardiac cells, ICl.Ca could be an important mediator of apoptosis[117]. But, information on the possible involvement of ICl.Ca in heart failure is currently very limited. It is reported that ICl.Camay play little, if any, role in the electrical remodeling of human end-stage failing heart[66,67,118].
Conclusions and future directions
Although the field of anion channels in cardiac physiology and pathophysiology lags significantly behind that of cation channels, the gap can now be narrowed with the recent identification of molecular entities responsible for cardiac Cl– channels[8], their genes mapped to specific human chromosomal locations[13] and the use of gene targeting and transgenic animals. Recent efforts not only at the cellular and molecular levels but also the isolated organ and whole animal levels have provided strong evidence that Cl– channels may play an important role in cardiac diseases, including arrhythmias, myocardial ischemia, hypertrophy, and congestive heart failure. Anion channels in the heart, therefore, may represent important novel targets for therapeutic agents against heart diseases.
Despite these exciting developments, further investigations of the cellular and molecular mechanisms by which the Cl– channel proteins function to impart a physiological or a pathophysiological phenotype may require a multitude of approaches for the assessment of the Cl– channel functions in healthy and diseased hearts. Although global knockout mice are invaluable experimental models and functional genomics remains a powerful approach to understanding the function of cardiac Cl– channels, several theoretical and practical problems should be considered. First, homologous recombination gene targeting is based on the assumption that targeting will result in specific loss of the gene’s product and will not directly affect the expression of other genes. In reality, however, even though the loss of the gene’s product can be verified, the upregulation of another gene in the vicinity of the targeting can occur[119] and may readily escape detection. Such upregulation could have an important effect on the observed phenotype. Second, a knockout may not always be a knockout[120] such as when the targeted gene is widely or ubiquitously expressed, when alternative splicing variants of the gene exist[121], and when functional channels are actually heteromultimeric and the structure might be associated with modulatory subunits, such as Barttin for ClC channels[122]. Accessory proteins may be involved in the determination of the stability of the channel complex in the membrane and in the modulation of biophysical, pharmaco-logical, and regulatory properties of the channel. Recent evidence suggests that Cl– channels, like cation chan-nels[52,123,124], may function as a multiprotein complex or functional module. A functional anion channel module may be a complex composed of the following: (a) pore forming subunit for ion transportation; (b) auxiliary subunits for modulating pore gating; and (c) proteins as second messengers tightly coupled to channel function. These proteins might be intimately linked to certain physiological functions and belong to the same subproteome. Manipulation of one gene in the subproteome may cause changes in other proteins of the same subproteome. Therefore, the functional consequences of disrupting the specific gene are very difficult to predict unless the changes in the entire subproteome are examined. Similar phenotypes can be attained from alternative protein pathways within cellular networks, which are influenced by disease, environmental, internal, and biochemical stimuli. Therefore, caution should be taken when conventional global gene knockout animals are used in functional studies. Alternatively, tissue-specific conditional or inducible knockout or knockin animal models may be more valuable in the phenotypic studies of specific genes by limiting the effect of upregulation or developmental compensation on the phenotype of manipulated genes. Many phenotypic changes may actually be a result of posttranslational changes caused by protein modifications such as phosphorylation or dephosphorylation. Therefore, it is clear that conventional functional genomics may provide only limited information on the functional module of multiprotein complexes. We are now facing the challenge of a major paradigm shift in the study of integrated anion channel functions. In the postgenomic era, the recent advances in the genome resources including genome-wide microarray profiling together with advancement in the application of functional proteomics and bioinformatics will certainly facilitate our understanding of the functions of anion channels in the cardiovascular system. It is feasible that anion channels may become novel targets for therapeutic approaches to the treatment of cardiovascular diseases.
Acknowledgements
The research in the Laboratory of Functional Genomics and Proteomics, Center of Biomedical Research Excellence and the Department of Pharmacology, University of Nevada, School of Medicine is supported by grants from the National Institutes of Health (R01-HL63914), National Center of Research Resources (NCRR, P20RR15581).
References
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 1999;79:917-1017.
- Marban E. Cardiac channelopathies. Nature 2002;415:213-8.
- Nattel S, Khairy P, Schram G. Arrhythmogenic ionic remodeling: adaptive responses with maladaptive consequences. Trends Cardiovasc Med 2001;11:295-301.
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature 2002;415:219-26.
- Nattel S. Human genetics: lost anchors cost lives. Nature 2003;421:587-90.
- Gross GJ, Peart JN. K. ATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol 2003;285:H921-30.
- O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res 2004;94:420-32.
- Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev 2000;80:31-81.
- Baumgarten CM, Clemo HF. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Prog Biophys Mol Biol 2003;82:25-42.
- Harvey RD. Cardiac chloride currents. News Phys Sci 1996;11:175-81.
- Hiraoka M, Kawano S, Hirano Y, Furukawa T. Role of cardiac chloride currents in changes in action potential characteristics and arrhythmias. Cardiovasc Res 1998;40:23-33.
- Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, et al. Functional significance of cell volume regulatory mechanisms. Physiol Rev 1998;78:247-306.
- Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev 1999;79:1317-72.
- Bahinski A, Nairn AC, Greengard P, Gadsby DC. Chloride conductance regulated by cyclic AMP-dependent protein kinase in cardiac myocytes. Nature 1989;340:718-21.
- Harvey RD, Hume JR. Autonomic regulation of a chloride current in heart. Science 1989;244:983-5.
- Nagel G, Hwang TC, Nastiuk KL, Nairn AC, Gadsby DC. The protein kinase A-regulated cardiac Cl– channel resembles the cystic fibrosis transmembrane conductance regulator. Nature 1992;360:81-4.
- Collier ML, Hume JR. Unitary chloride channels activated by protein kinase C in guinea pig ventricular myocytes. Circ Res 1995;76:317-24.
- Walsh KB, Long KJ. Properties of a protein kinase C-activated chloride current in guinea pig ventricular myocytes. Circ Res 1994;74:121-9.
- Duan D, Ye L, Britton F, Miller LJ, Yamazaki J, Horowitz B, et al. Purinoceptor-coupled Cl– channels in mouse heart: a novel, alternative pathway for CFTR regulation. J Physiol 1999;521:43-56.
- Levesque PC, Hume JR. ATPo but not cAMPi activates a chloride conductance in mouse ventricular myocytes. Cardiovasc Res 1995;29:336-43.
- Yamamoto-Mizuma S, Wang GX, Hume JR. P2Y purinergic receptor regulation of CFTR chloride channels in mouse cardiac myocytes. J Physiol 2004;556:727-37.
- Duan D, Ye L, Britton F, Horowitz B, Hume JR. A novel anionic inward rectifier in native cardiac myocytes. Circ Res 2000;86:E63-E71.
- Komukai K, Brette F, Orchard CH. Electrophysiological response of rat atrial myocytes to acidosis. Am J Physiol Heart Circ Physiol 2002;283:H715-24.
- Komukai K, Brette F, Pascarel C, Orchard CH. Electrophysiological response of rat ventricular myocytes to acidosis. Am J Physiol Heart Circ Physiol 2002;283:H412-22.
- Duan D, Fermini B, Nattel S. Sustained outward current observed after Ito1 inactivation in rabbit atrial myocytes is a novel Cl–current. Am J Physiol 1992;263:H1967-71.
- Duan D, Nattel S. Properties of single outwardly rectifying Cl– channels in heart. Circ Res 1994;75:789-95.
- Duan D, Fermini B, Nattel S. Alpha-adrenergic control of volume-regulated Cl– currents in rabbit atrial myocytes. Characterization of a novel ionic regulatory mechanism. Circ Res 1995;77:379-93.
- Duan D, Hume JR, Nattel S. Evidence that outwardly rectifying Cl– channels underlie volume-regulated Cl– currents in heart. Circ Res 1997;80:103-13.
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature 1997;390:417-21.
- Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J Gen Physiol 1999;113:57-70.
- Duan D, Zhong J, Hermoso M, Satterwhite CM, Rossow CF, Hatton WJ, et al. Functional inhibition of native volume-sensitive outwardly rectifying anion channels in muscle cells and Xenopus oocytes by anti-ClC-3 antibody. J Physiol 2001;531:437-44.
- Hermoso M, Satterwhite CM, Andrade YN, Hidalgo J, Wilson SM, Horowitz B, et al. ClC-3 is a fundamental molecular component of volume-sensitive outwardly rectifying Cl– channels and volume regulation in HeLa cells and Xenopus laevis oocytes. J Biol Chem 2002;277:40066-74.
- Wang GX, Hatton WJ, Wang GL, Zhong J, Yamboliev I, Duan D, et al. Functional effects of novel anti-ClC-3 antibodies on native volume-sensitive osmolyte and anion channels in cardiac and smooth muscle cells. Am J Physiol Heart Circ Physiol 2003;285:H1453-63.
- Yamamoto-Mizuma S, Wang GX, Liu LL, Schegg K, Hatton WJ, Duan D, et al. Altered properties of volume-sensitive osmolyte and anion channels (VSOACs) and membrane protein expression in cardiac and smooth muscle myocytes from Clcn3-/- mice. J Physiol 2004;557:439-56.
- Britton FC, Ohya S, Horowitz B, Greenwood IA. Comparison of the properties of CLCA1 generated currents and ICl(Ca) in murine portal vein smooth muscle cells. J Physiol 2002;539:107-17.
- Collier ML, Levesque PC, Kenyon JL, Hume JR. Unitary Cl- channels activated by cytoplasmic Ca2+ in canine ventricular myocytes. Circ Res 1996;78:936-44.
- Zygmunt AC, Gibbons WR. Properties of the calcium-activated chloride current in heart. J Gen Physiol 1992;99:391-414.
- Xu Y, Dong PH, Zhang Z, Ahmmed GU, Chiamvimonvat N. Presence of a calcium-activated chloride current in mouse ventricular myocytes. Am J Physiol Heart Circ Physiol 2002;283:H302-14.
- Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol 2005;67:221-40.
- Qu Z, Wei RW, Mann W, Hartzell HC. Two Bestrophins cloned from Xenopus laevis oocytes express Ca2+-activated Cl– currents. J Biol Chem 2003;278:49563-72.
- Qu Z, Fischmeister R, Hartzell C. Mouse Bestrophin-2 is a bona fide Cl– channel: identification of a residue important in anion binding and conduction. J Gen Physiol 2004;123:327-40.
- Qu Z, Hartzell C. Determinants of anion permeation in the second transmembrane domain of the mouse Bestrophin-2 chloride channel. J Gen Physiol 2004;124:371-82.
- Baumgarten CM, Fozzard HA. Intracellular chloride activity in mammalian ventricular muscle. Am J Physiol 1981;241:C121-9.
- Caille JP, Ruiz-Ceretti E, Schanne OF. Intracellular chloride activity in rabbit papillary muscle: effect of ouabain. Am J Physiol 1981;240:C183-8.
- Spitzer KW, Walker JL. Intracellular chloride activity in quiescent cat papillary muscle. Am J Physiol 1980;238:H487-93.
- Vaughan-Jones RD. Non-passive chloride distribution in mammalian heart muscle: micro-electrode measurement of the intracellular chloride activity. J Physiol (Lond) 1979;295:83-109.
- Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, et al. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation 1994;90:2534-9.
- Harvey RD, Clark CD, Hume JR. Chloride current in mammalian cardiac myocytes. Novel mechanism for autonomic regulation of action potential duration and resting membrane potential. J Gen Physiol 1990;95:1077-102.
- Matsuoka S, Ehara T, Noma A. Chloride-sensitive nature of the adrenaline-induced current in guinea-pig cardiac myocytes. J Physiol (Lond) 1990;425:579-98.
- Takano M, Noma A. Distribution of the isoprenaline-induced chloride current in rabbit heart. Pflugers Arch 1992;420:223-6.
- Yamawake N, Hirano Y, Sawanobori T, Hiraoka M. Arrhythmo-genic effects of isoproterenol-activated Cl– current in guinea-pig ventricular myocytes. J Mol Cell Cardiol 1992;24:1047-58.
- Ruiz PE, Ponce ZA, Schanne OF. Early action potential shortening in hypoxic hearts: role of chloride current(s) mediated by catecholamine release. J Mol Cell Cardiol 1996;28:279-90.
- Du XY, Sorota S. Cardiac swelling-induced chloride current depolarizes canine atrial myocytes. Am J Physiol 1997;272:H1904-16.
- Vandenberg JI, Bett GC, Powell T. Contribution of a swelling-activated chloride current to changes in the cardiac action potential. Am J Physiol 1997;273:C541-7.
- Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res 1999;42:270-83.
- Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res 1999;84:157-65.
- Hagiwara N, Masuda H, Shoda M, Irisawa H. Stretch-activated anion currents of rabbit cardiac myocytes. J Physiol (Lond) 1992;456:285-302.
- Browe DM, Baumgarten CM. Stretch of beta 1 integrin activates an outwardly rectifying chloride current via FAK and Src in rabbit ventricular myocytes. J Gen Physiol 2003;122:689-702.
- Browe DM, Baumgarten CM, Angiotensin II. AT1) receptors and NADPH oxidase regulate Cl– current elicited by β1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol 2004;124:273-87.
- Zygmunt AC, Gibbons WR. Calcium-activated chloride current in rabbit ventricular myocytes. Circ Res 1991;68:424-37.
- Verkerk AO, Tan HL, Ravesloot JH. Ca2+-activated Cl– current reduces transmural electrical heterogeneity within the rabbit left ventricle. Acta Physiol Scand 2004;180:239-47.
- Zygmunt AC. Intracellular calcium activates a chloride current in canine ventricular myocytes. Am J Physiol 1994;267:H1984-95.
- January CT, Fozzard HA. Delayed afterdepolarizations in heart muscle: mechanisms and relevance. Pharmacol Rev 1988;40:219-27.
- Verkerk AO, Veldkamp MW, Bouman LN, van Ginneken AC. Calcium-activated Cl– current contributes to delayed afterdepolariza-tions in single Purkinje and ventricular myocytes. Circulation 2000;101:2639-44.
- Verkerk AO, Veldkamp MW, de Jonge N, Wilders R, van Ginneken AC. Injury current modulates afterdepolarizations in single human ventricular cells. Cardiovasc Res 2000;47:124-32.
- Verkerk AO, Veldkamp MW, Baartscheer A, Schumacher CA, Klopping C, van Ginneken AC, et al. Ionic mechanism of delayed afterdepolarizations in ventricular cells isolated from human end-stage failing hearts. Circulation 2001;104:2728-33.
- Verkerk AO, Wilders R, Coronel R, Ravesloot JH, Verheijck EE. Ionic remodeling of sinoatrial node cells by heart failure. Circulation 2003;108:760-6.
- Wright AR, Rees SA. Targeting ischaemia−cell swelling and drug efficacy Trends Pharmacol Sci 1997;18:224-8. [published erratum appears in Trends Pharmacol Sci 1997; 18: 345].
- Frace AM, Maruoka F, Noma A. Control of the hyperpolarization-activated cation current by external anions in rabbit sino-atrial node cells. J Physiol (Lond) 1992;453:307-18.
- Nakajima T, Sugimoto T, Kurachi Y. Effects of anions on the G protein-mediated activation of the muscarinic K+ channel in the cardiac atrial cell membrane. Intracellular chloride inhibition of the GTPase activity of GK. J Gen Physiol 1992;99:665-82.
- Diaz RJ, Losito VA, Mao GD, Ford MK, Backx PH, Wilson GJ. Chloride channel inhibition blocks the protection of ischemic preconditioning and hypo-osmotic stress in rabbit ventricular myocardium. Circ Res 1999;84:763-75.
- Diaz RJ, Batthish M, Backx PH, Wilson GJ. Chloride channel inhibition does block the protection of ischemic preconditioning in myocardium. J Mol Cell Cardiol 2001;33:1887-9.
- Batthish M, Diaz RJ, Zeng HP, Backx PH, Wilson GJ. Pharmacological preconditioning in rabbit myocardium is blocked by chloride channel inhibition. Cardiovasc Res 2002;55:660-71.
- Heusch G, Liu GS, Rose J, Cohen MV, Downey JM. No confirmation for a causal role of volume-regulated chloride channels in ischemic preconditioning in rabbits. J Mol Cell Cardiol 2000;32:2279-85.
- Bozeat ND, Dwyer L, Ye L, Yao T, Duan D. The role of ClC-3 chloride channels in early and late ischemic preconditioning in mouse heart. FASEB J 2005;32. in press.
- Mizoguchi K, Maeta H, Yamamoto A, Oe M, Kosaka H. Amelioration of myocardial global ischemia/reperfusion injury with volume-regulatory chloride channel inhibitors in vivo. Transplantation 2002;73:1185-93.
- Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol 2001;280:H2313-20.
- Lemonnier L, Shuba Y, Crepin A, Roudbaraki M, Slomianny C, Mauroy B, et al. Bcl-2-dependent modulation of swelling-activated Cl– current and ClC-3 expression in human prostate cancer epithelial cells. Cancer Res 2004;64:4841-8.
- Wei L, Xiao AY, Jin C, Yang A, Lu ZY, Yu SP. Effects of chloride and potassium channel blockers on apoptotic cell shrinkage and apoptosis in cortical neurons. Pflugers Arch 2004;448:325-34.
- Sheppard DN, Welsh MJ. Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J Gen Physiol 1992;100:573-91.
- Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl– channels expressed in a murine cell line. J Physiol (Lond) 1997;503:333-46.
- Yamazaki J, Hume JR. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated, Ca2+-activated Cl– channels in mammalian cardiac myocytes. Circ Res 1997;81:101-9.
- Lochner A, Genade S, Tromp E, Podzuweit T, Moolman JA. Ischemic preconditioning and the beta-adrenergic signal transduction pathway. Circulation 1999;100:958-66.
- Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, et al. Formation of protein kinase C (epsilon)-Lck signaling modules confers cardio-protection. J Clin Invest 2002;109:499-507.
- Yamazaki J, Britton F, Collier ML, Horowitz B, Hume JR. Regulation of recombinant cardiac cystic fibrosis transmembrane conductance regulator chloride channels by protein kinase C. Biophys J 1999;76:1972-87.
- Chen H, Liu LL, Ye LL, McGuckin C, Tamowski S, Scowen P, et al. Targeted inactivation of cystic fibrosis transmembrane conductance regulator chloride channel gene prevents ischemic preconditioning in isolated mouse heart. Circulation 2004;110:700-4.
- Ye L, Ge ZD, Liu L, Murray K, Hatton WJ, Auchampach JA, et al. In vivo study of the protective role of CFTR chloride channels in ischemic preconditioning. FASEB J 2003;174:A106.
- Boujaoude LC, Bradshaw-Wilder C, Mao C, Cohn J, Ogretmen B, Hannun YA, et al. Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J Biol Chem 2001;276:35258-64.
- Karliner JS. Lysophospholipids and the cardiovascular system. Biochim Biophys Acta 2002;1582:216-21.
- Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, et al. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J 2003;22:1981-9.
- Barriere H, Poujeol C, Tauc M, Blasi JM, Counillon L, Poujeol P. CFTR modulates programmed cell death by decreasing intracellular pH in Chinese hamster lung fibroblasts. Am J Physiol Cell Physiol 2001;281:C810-24.
- Valverde MA, Vazquez E, Munoz FJ, Nobles M, Delaney SJ, Wainwright BJ, et al. Murine CFTR channel and its role in regulatory volume decrease of small intestine crypts. Cell Physiol Biochem 2000;10:321-8.
- Piper HM, Meuter K, Schafer C. Cellular mechanisms of ischemia-reperfusion injury. Ann Thorac Surg 2003;75:S644-8.
- Piper HM, Abdallah Y, Schafer C. The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res 2004;61:365-71.
- Li GR, Du XL, Siow YL. O K, Tse HF, Lau CP. Calcium-activated transient outward chloride current and phase 1 repolarization of swine ventricular action potential. Cardiovasc Res 2003;58:89-98.
- Li GR, Sun H, To J, Tse HF, Lau CP. Demonstration of calcium-activated transient outward chloride current and delayed rectifier potassium currents in Swine atrial myocytes. J Mol Cell Cardiol 2004;36:495-504.
- Verkerk AO, Wilders R, Zegers JG, van Borren MM, Ravesloot JH, Verheijck EE. Ca2+-activated Cl– current in rabbit sinoatrial node cells. J Physiol 2002;540:105-17.
- Zygmunt AC, Goodrow RJ, Weigel CM. INa(Ca) and ICl(Ca) contribute to isoproterenol-induced delayed after depolarizations in midmyocardial cells. Am J Physiol 1998;275:H1979-92.
- Laser M, Willey CD, Jiang W, Cooper G, Menick DR, Zile MR, et al. Integrin activation and focal complex formation in cardiac hypertrophy. J Biol Chem 2000;275:35624-30.
- Weber KT, Sun Y, Guarda E. Structural remodeling in hypertensive heart disease and the role of hormones. Hypertension 1994;23:869-77.
- Colucci WS. Molecular and cellular mechanisms of myocardial failure. Am J Cardiol 1997;80:15L-25L.
- Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res 1999;84:157-65.
- Clemo HF, Baumgarten CM. Protein kinase C activation blocks ICl(swell) and causes myocyte swelling in a rabbit congestive heart failure model. Circulation 1998;98:I-695.
- Clemo HF, Danetz JS, Baumgarten CM. Does ClC-3 modulate cardiac cell volume? Biophys J 1999;76:A203.
- Clemo HF, Rana J, Vaida AM, Tseng GN, Higgins RS, Baumgarten CM. Chronic activation of ICl,swell in canine infarction model supressess inducibility of early afterdepolarizations. Circulation 2001;104:II-624.
- Patel DG, Higgins RS, Baumgarten CM. Swelling-activated Cl current, ICl,swell, is chronically activated in diseased human atrial myocytes. Biophys J 2003;84:233a.
- Benitah JP, Gomez AM, Delgado C, Lorente P, Lederer WJ. A chloride current component induced by hypertrophy in rat ventricular myocytes. Am J Physiol 1997;272:H2500-6.
- De Mello WC. Heart failure: how important is cellular sequestration? The role of the renin-angiotensin-aldosterone system. J Mol Cell Cardiol 2004;37:431-8.
- Liu L, Ye L, McGuckin C, Hatton WJ, Duan D. Disruption of Clcn3 gene in mice facilitates heart failure during pressure overload. J Gen Physiol 2003;122:76.
- Wondergem R, Gong W, Monen SH, Dooley SN, Gonce JL, Conner TD, et al. Blocking swelling-activated chloride current inhibits mouse liver cell proliferation. J Physiol 2001;532:661-72.
- Duffy SM, Leyland ML, Conley EC, Bradding P. Voltage-dependent and calcium-activated ion channels in the human mast cell line HMC-1. J Leukoc Biol 2001;70:233-40.
- Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H. Expression of voltage-gated chloride channels in human glioma cells. J Neurosci 2003;23:5572-82.
- Paul S. Ventricular remodeling. Crit Care Nurs Clin North Am 2003;15:407-11.
- Wong KR, Trezise AE, Crozatier B, Vandenberg JI. Loss of the normal epicardial to endocardial gradient of cftr mRNA expression in the hypertrophied rabbit left ventricle. Biochem Biophys Res Commun 2000;278:144-9.
- Davies WL, Vandenberg JI, Sayeed RA, Trezise AE. Post-transcriptional regulation of the cystic fibrosis gene in cardiac development and hypertrophy. Biochem Biophys Res Commun 2004;319:410-8.
- Houser SR, Piacentino V III, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol 2000;32:1595-607.
- Elble RC, Pauli BU. Tumor suppression by a proapoptotic calcium-activated chloride channel in mammary epithelium. J Biol Chem 2001;276:40510-7.
- Verkerk AO, Tan HL, Kirkels JH, Ravesloot JH. Role of Ca2+-activated Cl- current during proarrhythmic early afterdepolarizations in sheep and human ventricular myocytes. Acta Physiol Scand 2003;179:143-8.
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999;292:797-817.
- London B. A knockout may not always be a knockout. Circulation 2000;102:E122.
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev 2002;82:503-68.
- Estevez R, Boettger T, Stein V, Birkenhager R, Otto E, Hildebrandt F, et al. Barttin is a Cl– channel beta-subunit crucial for renal Cl– reabsorption and inner ear K+ secretion. Nature 2001;414:558-61.
- Ardehali H, Chen Z, Ko Y, Mejia-Alvarez R, Marban E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc Natl Acad Sci USA 2004;101:11880-5.
- Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, et al. Formation of protein kinase C(epsilon)-Lck signaling modules confers cardio-protection. J Clin Invest 2002;109:499-507.
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 2002;415:287-94.