
Nitric oxide: a newly discovered function on wound healing
Introduction
Wound repair is a well orchestrated and highly coordinated process that includes a series of overlapping phases: inflammation, cell proliferation, matrix deposition, and tissue remodeling. This involves a complex, dynamic series of events including clotting, inflammation, granulation tissue formation, epithelialization, neovascularization, collagen synthesis, and wound contraction[1]. Loss of a functional healing process could lead to severe disabilities. Accord-ingly, chronic, non-healing wound conditions represent a situation of major clinical importance. The series of pathological changes associated with several diseases ultimately leads to severely disturbed wound healing conditions[2]. Among those, the most prominent chronic wound impairments include decubitus or pressure ulcers, venous ulcers, and diabetic ulcers. The advent of molecular and cellular biology and the use of different modeling systems, most notably genetically engineered animals, have greatly extended our knowledge of wound repair. Inflammation, re-epitheliali-zation, and granulation tissue formation are driven in part by a complex mixture of growth factors and cytokines, which are released coordinately into the wounds[1,2]. Besides these protein-type factors and mitogens, evidence is emerging for the important role of small diffusible molecules such as nitric oxide (NO) in wound repair[3]. In this review, we summarize the current knowledge of the modulating functions of NO on wound repair.
Chemistry and biosynthesis of nitric oxide
NO is a highly diffusible intercellular signaling molecule implicated in a wide range of biological effects. It is generated by the enzyme nitric oxide synthase (NOS), which catalyzes the conversion of L-arginine to L-citrulline[4]. Three NOS isoforms have been characterized, each encoded by different chromosomes. Two enzyme isoforms are constitutively expressed (endothelial and neuronal NOS), whereas one isoform is an inducible enzyme (iNOS), initially found in macrophages. All three NOS isoforms exist in their active form of homodimers of two domains: a C-terminal reductase domain, and an N-terminal oxygenase domain with molecular masses of approximately 135 kDa (eNOS), 150–160 kDa (nNOS), and 130 kDa (iNOS)[4]. The reductase domain contains binding sites for one molecule each of nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), and flavin dinucleotide (FAD), in close homology with cytochrome P-450 reductase, whereas the oxygen domain binds heme, the essential cofactor tetrahydrobiopterin (BH4), and the substrate L-arginine[4]. Between these two regions lies the calmodulin (CaM) binding site, which plays a key role in both the structure and function of the enzyme. The constitutive isoforms (eNOS and nNOS) are permanently active, generating low concentrations of NO (in nmol/L range). Their enzymatic activities are regulated by intracellular calcium fluxes or exogenous calmodulin. The expression, transcription, and function of the iNOS is induced by a variety of cytokines, growth factors, and inflammatory stimuli on target cells which lead to the release of much higher levels of NO (in µmol/L range), which is involved in host immune response.
All three NOS isoforms are expressed in skin tissue[3]. Expression of nNOS has been observed in keratinocytes and melanocytes; eNOS can be detected in keratinocytes of the basal epidermal layer, dermal fibroblasts, endothelial capillaries, and eccrine glands; and iNOS can be induced in keratinocytes, fibroblasts, Langerhans, and endothelial cells. Accordingly, NO participates in the regulation of skin homeostatic functions such as circulation, sunburn erythema, and maintenance of the protective barrier against micro-organisms.
Nitric oxide and wound healing
L-Arginine, the substrate for NOS, was first noted to enhance wound healing in 1978[5]. Subsequently, dietary L-arginine intake has been shown to improve collagen deposition and wound strength in both animals and humans[6–8]. This effect of L-arginine may be due in part to its conversion to L-ornithine through the action of arginase, an enzyme that may compete with NOS for L-arginine and thereby help regulate NO production during wound healing[9]. However, the finding that L-arginine intake does not improve collagen deposition in iNOS-deficient mice to the same extent as in wild-type littermates implicates that part of L-arginine’s effect involves NO directly[10].
Accumulating evidence indicates that NO plays a key role in normal wound repair (Figure 1)[11–18]. Production of nitrite (NO2) and nitrate (NO3), the stable NO metabolites, are elevated early in the fluid of subcutaneous wounds[11], and urinary nitrate excretion increases in a sustained manner after excisional wounding[12]. Furthermore, the presence of nitrite and nitrate is directly correlated with collagen deposition within the wound and in dermal fibroblasts, suggesting that NO synthesis is critical for wound collagen accumulation and acquisition of mechanical strength[11,13,14]. All three NOS isozymes are involved in the wound healing process. Both iNOS and nNOS mRNA and protein expression are increased in cutaneous wounds[15,16]. Our recent findings demonstrate that there is a significant increase of cutaneous eNOS protein expression as well as constitutive NOS enzymatic activity after excisional wounding in normal mice[17]. Consequently, an NO deficiency directly contributes to wound healing impairment. Inhibition of NOS by competitive inhibitors, either applied to the wound surface[18] or given systemically[11], decreases collagen deposition and breaking strength of incisional wounds and impairs the healing.
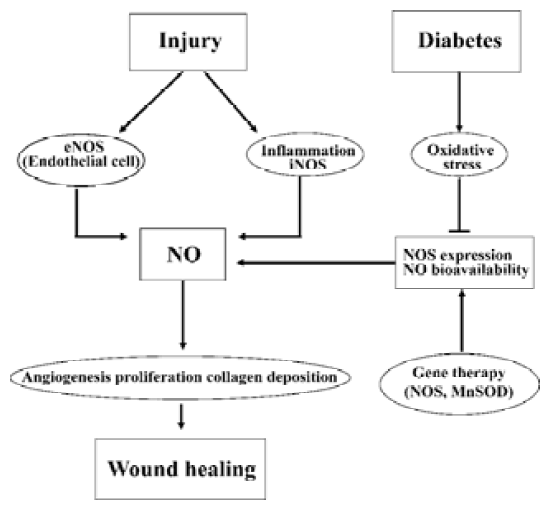
Consistent with these findings, studies with targeted disruption of NOS genes have revealed that the excisional wound closure is delayed by 30% in both eNOS and iNOS knockout mice compared to their wild-type littermates[19,20]. Conversely, adenoviral vector-mediated gene transfer of iNOS to the wound site of iNOS knockout mice completely reversed the delayed healing[20].
Finally, there are strong correlations between reduced cutaneous NO levels and impaired wound healing under disease conditions such as diabetes[11,14,17,21], malnutrition[13], and chronic steroid treatment[18]. Diabetic wound healing impairment is one of the most well-known chronic wound situations. Studies of ours and others demonstrate that cutaneous eNOS expression, constitutive NOS activity, and/or NO levels are significantly decreased in streptozotocin (STZ)-induced type 1 diabetic animals[11,14,17,21]. In fact, our findings indicate that the augmented cutaneous eNOS protein expression and constitutive NOS activity observed in normal animals in the healing process are absent in the type 1 diabetic mice[17]. These findings suggest that impairment of wound-induced endogenous NOS expression and NOS activity is responsible for reduced cutaneous NO bioavail-ability in type 1 diabetic animals (Figure 1). In agreement with the above notion, cutaneous gene therapy of eNOS or manganese superoxide dismutase (MnSOD) restored eNOS protein and NO levels and accelerated the wound healing rate in STZ-induced diabetic mice[17]. Similarly, the NO donor molsidomine (N-ethoxycarbomyl-3-morpholinyl-sidnonimine) or NO releasing poly (vinyl alcohol) hydrogel dressings are also shown to partially restore such healing impairment in STZ-induced diabetic rats[22,23]. Collectively, impairment of skin NO function represents an important factor for delayed wound healing in diabetes and strategies aimed at restoring cutaneous NO bioavailability with NO donors or NOS gene therapy may serve as effective means for diabetic wound healing.
Mechanisms of nitric oxide on wound healing
NO and angiogenesis Angiogenesis, the process of forming new microvessels, is an important component of normal wound repair. NO plays a central role in this process[24] as it increases angiogenesis in ischemic murine tissues[25]. Conversely, NOS inhibitors impair angiogenesis in granulation tissue during gastric ulcer healing[26] and suppress capillary organization in vitro[27].
NO is also vital to the activity of pro-angiogenic cytokines. Vascular endothelial growth factor (VEGF) is a potent angiogenic factor which involves the modulation of NO generation[28]. VEGF increases NO production via upregulation of eNOS[29,30]. Conversely, the angiogenic effect of VEGF also depends on NO as pharmacological blockade of NOS prevent both VEGF-induced endothelial cell proliferation and mitogen-activated protein (MAP) kinase[31]. VEGF-stimulated endothelial cell migration, decreased adhesion, and organization are also dependent on NO[32,33]. Keratinocytes are the major source of VEGF expression upon cytokine stimulation[34], which is blocked by iNOS inhibitors both in vitro and in vivo[35]. NO has also been shown to downregulate protein kinase C (PKC)-induced VEGF expression in smooth muscle cells by interfering with the binding of AP-1[36] and to participate in the conversion of VEGF from an inert to an angiogenic form[37]. Interestingly, NO is also involved in VEGF-independent angiogenesis mechanisms. Evidence includes the role of NO in monocyte-induced angiogenesis induced by monocytes[38], substance P[39], and transforming growth factor (TGF)-β1[40]. Taken together, these studies suggest a vital role of NO in post-wound angiogenesis.
NO and inflammation NO has been shown to modulate chemoattractant cytokines that initiate post-wound inflammation, including interleukin (IL)-8[41], TGF-β1[42], monocytes, and neutrophils[43] that contribute to wound chemoattraction. Once monocytes and neutrophils are attracted to the site of a wound, they are activated and begin to produce TNF-α and IL-1, both of which are implicated in wound healing[33]. Because IL-1 is a potent chemoattrac-tant for keratinocytes, the modulation of IL-1 by NO may usher keratinocyte recruitment, proliferation, and differentiation. Taken together, NO modulation of inflammation-associated cytokines may affect the inflammatory phase of wound healing.
NO and cell proliferation, differentiation, and apopto-sis NO affects proliferation, differentiation, and apoptosis in a number of cell types involved in wound healing. The iNOS inhibitor Nω-imino ethyl L-lysine (L-NIL) has been found to decrease proliferation in keratinocytes at the wound edge[44]. Indeed, treatment of murine wounds with L-NIL leads to delayed re-epithelialization with atrophied hyper-proliferative epithelium seen at the wound edge[45]. Conver-sely, the NO donor sodium nitroprusside (SNP) significantly increases fetal bovine serum-induced thymidine incorporation into the DNA of human dermal fibroblasts and enhances fibroblast growth factor- or platelet-drived growth factor-induced DNA synthesis[46]. Furthermore, low levels of NO increase keratinocyte proliferation in vitro[44], an effect that is mimicked by 8-bromo-cGMP[33], an analog of NO second messenger cGMP. NO also modulates keratinocyte apoptosis induced by irradiation of keratinocytes with ultraviolet B light as addition of NOS inhibitors to irradiated keratinocytes increases apoptosis, an effect that is reversed by the NO donor S-nitroso-penicillamine[47]. It appears that both inducible and constitutive NOS are involved in this process. Furthermore, NO has been shown to stimulate the proliferation of endothelial cells, protect endothelial cells from apoptosis, and mediate VEGF production[34]. These effects of NO on endothelial cells may also be related to another facet of wound healing, namely angiogenesis. In contrast, NO may also affect fibroblast proliferation. For instance, NO donor SNAP has been reported to decrease the proliferation of normal dermal fibroblasts in rats[48] while increase their proliferation in mice[49], even though the reasons for such discrepancy are not clear. Altogether, the above studies suggest that NO affect the proliferative phase of wound healing.
NO and matrix deposition and remodeling The final phases of healing require increased collagen synthesis and deposition, and a link between NO and collagen deposition has been described[11]. In most studies, treatment with NO donors, dietary L-arginine, or iNOS overexpression via gene therapy increased the collagen content of experimental wounds[10,11,50,51]. Indeed, treatment with a NO donor has been shown to increase collagen formation in fibroblasts derived from both normal and wound skin, which was decreased following NOS inhibition[51]. The effect of NO may primarily be due to the posttranslational enhancement of collagen synthesis rather than de novo transcription of the relevant collagen genes[51].
Mechanisms of wound nitric oxide dysfunction
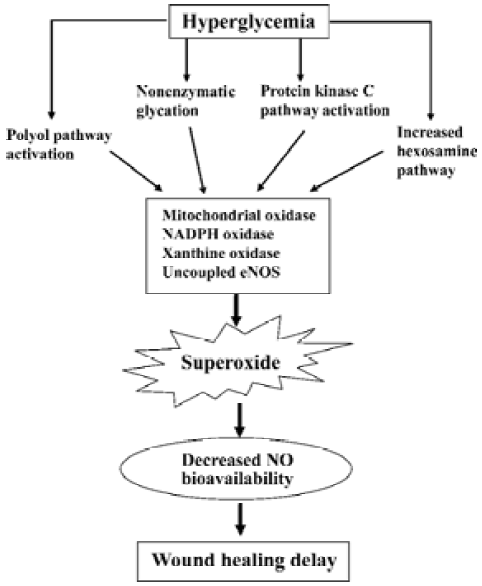
Although impaired NO function contributes to delayed wound healing in diabetes, the mechanisms of cutaneous NO dysfunction in this setting is unclear. In diabetes, causative factors for hyperglycemia-induced organ damage include the activation of the polyol pathway, nonenzymatic glycation, activation of PKC pathway, and increased hexosamine pathway flux[52]. However, previously there was no apparent common element linking these mechanisms. Recent studies suggest that these different mechanisms may be linked by a single cellular process: an overproduction of superoxide induced by sustained hyperglycemia[53]. Sustained hyperglycemia is known to increase vascular superoxide levels, resulting in cardiovascular dysfunction[54]. Superoxide produced in the vasculature rapidly inactivates NO and thus reduces its bioavailability in diabetic vasculature[55]. Independent strategies aimed at reducing superoxide levels have been shown to prevent high glucose-induced PKC activation, formation of advanced glycation end-products (AGEs), sorbitol accumulation, and NFκB activation, resulting in improvement of endothelium-dependent NO-mediated vasodilation[53,54]. These results indicate that increased superoxide levels are a key factor in vascular NO dysfunction in diabetes. However, whether sustained hyperglycemia increases cutaneous O2– levels and the mechanisms by which cutaneous NO levels are decreased in diabetes is unknown. Our recent studies demonstrate that glucose concentration-dependently increases superoxide levels in normal mouse skin and there is a marked increase of cutaneous superoxide levels in streptozotocin-induced type 1 diabetic mice[17]. Furthermore, cutaneous gene therapy of MnSOD significantly reduced superoxide and increased NO levels, resulting in accelerated wound healing in this model. These results provide the direct evidence that increased cutaneous superoxide contributes to reduced NO bioavailability and wound healing impairment in diabetes (Figure 2).
Future directions
Although a central role for several protein-type growth factors and mitogens on wound repair has been well-established for many years, the application of these factors in the treatment of wound healing has not provided a breakthrough in the clinical arena[1,2]. One possible reason for the failure of markedly accelerating closure of chronic wounds may be due to increased protease activities in the wound fluids, which may impair the ability of endogenous and exogenously applied growth factor proteins to stimulate healing. In contrast, NO may represent a novel target molecule to circumvent these difficulties. Because NO is a short-lived gas molecule, maintaining an effective level of NO at the wound site is an obvious problem for clinical therapy. In recent studies we have demonstrated that gene therapy of NOS or SOD is effective in restoring cutaneous NO levels and accelerating wound healing in diabetic mice[17]. Gene therapy strategies aimed at increasing NO or reducing superoxide levels may represent an effective means of reversing cutaneous NO deficiency at the wound site for healing refractory wounds in diabetes and other diseases. Future preclinical studies are warranted to optimize the designs and regimens before clinical trails can be conducted and the ultimate translation of basic science to the clinical settings for human gene therapy.
Acknowledgements
This work was supported in part by the Juvenile Diabetes Research Foundation (JDRF) Innovative Grant 5-2001-311, American Diabetes Association (ADA) Regular Research Award 7-01-RA-10, American Heart Association (AHA) Grants 9806347X, 0130537Z and 0455594Z, and the Intramural Research Grants Program (IRGP) grant #41140 of the Michigan State University (all to Dr Alex F CHEN).
References
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med 1999;341:738-46.
- Bello YM, Phillips TJ. Recent advances in wound healing. JAMA 2000;283:716-8.
- Frank S, Kampfer H, Wetzler C, Pfeilschifter J. Nitric oxide drives skin repair: novel functions of an established mediator. Kidney Int 2002;61:882-8.
- Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol 1995;57:707-36.
- Seifter E, Rettura G, Barbul A, Levenson SM. Arginine: an essential amino acid for injured rats. Surgery 1978;84:224-30.
- Barbul A, Lazarou SA, Efron DT, Wasserkrug HL, Efron G. Arginine enhances wound healing and lymphocyte immune responses in humans. Surgery 1990;108:331-6.
- Kirk SJ, Hurson M, Regan MC, Holt DR, Wasserkrug HL, Barbul A. Arginine stimulates wound healing and immune function in elderly human beings. Surgery 1993;114:155-9.
- Arbss MA, Ferrando JM, Vidal J, Quiles MT, Huguet P, Castells J, et al. Early effects of exogenous arginine after the implantation of prosthetic material into the rat abdominal wall. Life Sci 2000;67:2493-512.
- Albina JE, Mills CD, Henry WL Jr, Caldwell MD. Temporal expression of different pathways of L-arginine metabolism in healing wounds. J Immunol 1990;144:3877-80.
- Shi HP, Efron DT, Most D, Tantry US, Barbul A. Supplemental dietary arginine enhances wound healing in normal but not inducible nitric oxide synthase knockout mice. Surgery 2000;128:374-8.
- Schaffer MR, Tantry U, Gross SS, Wasserburg HL, Barbul A. Nitric oxide regulates wound healing. J Surg Res 1996;63:237-40.
- Smith DJ, Dunphy MJ, Strag LN, Marletta MA. The influence of wound healing on urinary nitrate levels in rats. Wounds 1991;3:50-8.
- Schaffer MR, Tantry U, Ahrendt GM, Wasserburg HL, Barbul A. Acute protein-calorie malnutrition impairs wound healing: a possible role of decreased wound nitric oxide synthesis. J Am Coll Surg 1997;184:37-43.
- Schaffer MR, Tantry U, Efron PA, Ahrendt GM, Thornton FJ, Barbul A. Diabetes-impaired healing and reduced wound nitric oxide synthesis: a possible pathophysiologic correlation. Surgery 1997;121:513-9.
- Frank S, Madlener M, Pfeilschifter J, Werner S. Induction of inducible nitric oxide synthase and its corresponding tetrahydrobiopterin-cofactor-synthesizing enzyme GTP-cyclohydrolase I during cutaneous wound repair. J Invest Dermatol 1998;111:1058-64.
- Boissel JP, Ohly D, Bros M, Godtel-Armbrust U, Forstermann U, Frank S. The neuronal nitric oxide synthase is upregulated in mouse skin repair and in response to epidermal growth factor in human HaCaT keratinocytes. J Invest Dermatol 2004;123:132-9.
- Luo JD, Wang YY, Fu W, Wu J, Chen AF. Gene therapy of eNOS and MnSOD restores delayed wound healing in type 1 diabetic mice. Circulation 2004;110:2484-93.
- Bulgrin JP, Shabani M, Chakravarthy D, Smith DJ. Nitric oxide synthesis is suppressed in steroid-impaired and diabetic wounds. Wounds 1995;7:48-57.
- Lee PC, Salyapongse AN, Bragdon GA, Shears LL, Watkins SC, Edington HDJ, et al. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am J Physiol 1999;277:H1600-8.
- Yamasaki K, Edington HDJ, McClosky C, Tzeng E, Lizonova A, Kovesdi I, et al. Reversal of impaired wound repair in iNOS-deficient mice by topical adenoviral-mediated iNOS gene transfer. J Clin Invest 1998;101:967-71.
- Stallmeyer B, Anhold M, Wetzler C, Kahlina K, Pfeilschifter J, Frank S. Regulation of eNOS in normal and diabetes-impaired skin repair: implications for tissue regeneration. Nitric Oxide 2002;6:168-77.
- Witte MB, Kiyama T, Barbul A. Nitric oxide enhances experimental wound healing in diabetes. Br J Surg 2002;89:1594-601.
- Masters KS, Leibovich SJ, Belem P, West JL, Poole-Warren LA. Effects of nitric oxide releasing poly(vinyl alcohol) hydrogel dressings on dermal wound healing in diabetic mice. Wound Repair Regen 2002;10:286-94.
- Donnini S, Ziche M. Constitutive and inducible nitric oxide synthase: role in angiogenesis. Antioxid Redox Signal 2002;4:817-23.
- Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 1998;101:2567-78.
- Konturek SJ, Brzozowski T, Majka J, Pytko-Polonczyk J, Stachura J. Inhibition of nitric oxide synthase delays healing of chronic gastric ulcers. Eur J Pharmacol 1993;239:215-7.
- Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest 1997;100:3131-9.
- Zhang R, Wang L, Zhang L, Chen J, Zhu Z, Zhang Z, et al. Nitric oxide enhances angiogenesis via the synthesis of vascular endothelial growth factor and cGMP after stroke in the rat. Circ Res 2003;92:308-13.
- Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol 1998;274:H1054-8.
- Gelinas DS, Bernatchez PN, Rollin S, Bazan NG, Sirois MG. Immediate and delayed VEGF-mediated NO synthesis in endothelial cells: role of PI3K, PKC and PLC pathways. Br J Pharmacol 2002;137:1021-30.
- Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, et al. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest 1997;99:2625-34.
- Morbidelli L, Chang CH, Douglas JG, Granger HJ, Ledda F, Ziche M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am J Physiol 1996;270:H411-5.
- Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric oxide and wound repair: role of cytokines? Nitric Oxide 2002;7:1-10.
- Frank S, Kampfer H, Wetzler C, Pfeilschifter J. Nitric oxide drives skin repair: novel functions of an established mediator. Kidney Int 2002;61:882-8.
- Frank S, Stallmeyer B, Kampfer H, Kolb N, Pfeilschifter J. Nitric oxide triggers enhanced induction of vascular endothelial growth factor expression in cultured keratinocytes (HaCaT) and during cutaneous wound repair. FASEB J 1999;13:2002-14.
- Tsurumi Y, Murohara T, Krasinski K, Chen D, Witzenbichler B, Kearney M, et al. Reciprocal relation between VEGF and NO in the regulation of endothelial integrity. Nat Med 1997;3:879-86.
- Xiong M, Elson G, Legarda D, Leibovich SJ. Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol 1998;153:587-98.
- Leibovich SJ, Polverini PJ, Fong TW, Harlow LA, Koch AE. Production of angiogenic activity by human monocytes requires an L-arginine/nitric oxide-synthase-dependent effector mechanism. Proc Natl Acad Sci USA 1994;91:4190-4.
- Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest 1994;94:2036-44.
- Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA 1986;83:4167-71.
- Andrew PJ, Harant H, Lindley IJ. Nitric oxide regulates IL-8 expression in melanoma cells at the transcriptional level. Biochem Biophys Res Commun 1995;214:949-56.
- Malik AA, Radhakrishnan N, Reddy K, Smith AD, Singhal PC. Tubular cell-Escherichia coli interaction products modulate migration of monocytes through generation of transforming growth factor-beta and macrophage-monocyte chemoattractant protein-1. J Endourol 2002;16:599-603.
- Belenky SN, Robbins RA, Rubinstein I. Nitric oxide synthase inhibitors attenuate human monocyte chemotaxis in vitro. J Leukoc Biol 1993;53:498-503.
- Stallmeyer B, Kampfer H, Kolb N, Pfeilschifter J, Frank S. The function of nitric oxide in wound repair: inhibition of inducible nitric oxide-synthase severely impairs wound reepithelialization. J Invest Dermatol 1999;113:1090-8.
- Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest 2000;117:1162-72.
- Dhaunsi GS, Ozand PT. Nitric oxide promotes mitogen-induced DNA synthesis in human dermal fibroblasts through cGMP. Clin Exp Pharmacol Physiol 2004;31:46-9.
- Seo SJ, Choi HG, Chung HJ, Hong CK. Time course of expression of mRNA of inducible nitric oxide synthase and generation of nitric oxide by ultraviolet B in keratinocyte cell lines. Br J Dermatol 2002;147:655-62.
- Witte MB, Thornton FJ, Efron DT, Barbul A. Enhancement of fibroblast collagen synthesis by nitric oxide. Nitric Oxide 2000;4:572-82.
- Schwentker A, Billiar TR. Inducible nitric oxide synthase: from cloning to therapeutic applications. World J Surg 2002;26:772-8.
- Thornton FJ, Schaffer MR, Witte MB, Moldawer LL, MacKay SL, Abouhamze A, et al. Enhanced collagen accumulation following direct transfection of the inducible nitric oxide synthase gene in cutaneous wounds. Biochem Biophys Res Commun 1998;246:654-9.
- Schaffer MR, Efron PA, Thornton FJ, Klingel K, Gross SS, Barbul A. Nitric oxide, an autocrine regulator of wound fibroblast synthetic function. J Immunol 1997;158:2375-81.
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813-20.
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemia damage. Nature 2000;404:787-90.
- Kim YK, Lee MS, Son SM, Kim IJ, Lee WS, Rhim BY, et al. Vascular NADH oxidase is involved in impaired endothelium-dependent vasodilation in OLETF rats, a model of type 2 diabetes. Diabetes 2002;51:522-7.
- Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 2001;88:E14-22.