
Pharmacokinetics and tissue distribution of 5-fluorouracil encapsulated by galactosylceramide liposomes in mice1
Introduction
5-Fluorouracil (5-Fu) is a chemotherapeutic drug that is used widely for the treatment of malignant cancers, and is usually the first choice of drug for the treatment of hepatic cellular cancer. High doses of this drug are currently administered, mostly by continuous infusion, over 5 to 21 d[1]. But its use has been limited by its systemic toxicities, which have severe gastrointestinal toxicities, hematologic side effects, and severe disturbance in bone marrow[2]. Moreover, 5-Fu has a serum half-life of only 15 min, further limiting its usefulness[3]. Several studies have reported a relationship between 5-Fu plasma levels and its toxicity, and the response to treatment. 5-Fu is often monitored in serum by pharmacokinetic follow-up with liquid chromatography[4].
In order to maximize the therapeutic effect of 5-Fu and minimize its adverse effects, microsphere, liposome, implants, and nanoparticles were made[5–8]. But they were all passive for targeting preparations. Galactosylceramide (GC), as a novel membrane material, was adopted to envelop 5-Fu and form liposomes. This kind of 5-Fu preparation has not been previously reported. The purpose of this study was to measure the pharmacokinetic parameters and tissue distribution of 5-Fu-GCL in mice, which should supply evidence for its long effect and active targeting.
Materials and methods
Drugs and reagents 5-Fluorouracil encapsulated by galactosylceramide liposomes (5-Fu-GCL) (Batch N
Apparatus Shimadzu LC-10A HPLC equipment consisted of LC-10A pumps, SCL-6A system controller, SPD-6AV ultraviolet spetrophotometric detector, ODS-C18 column(150 mm×4.6 mm) (Shimadzu Co, Japan), and V4.0+ software on a computer.
Animals Balb/c mice (male and female, 18–22 g) in the study were provided by the Experimental Animal Center of Anhui Medical University (certifcate N
Liposome preparation 5-Fu was encapsulated in liposomes made of galactocerebroside, Lecithin Granules and cholesterin in a molar ratio of 6:3:1. Liposomes were prepared by combined calcium-induced fusion with reverse evaporation method. In brief, phospholipids were dissolved in 0.7 mL of carbinol and chloroform compound with 2:1 volume and subjected to rotary evaporation until the lipid membrane formed. Then 1.5 mL of chloroform, 250 µL of 5-Fu (10 g/L) and 250 µL of ultrasonic buffer were added to the dried phospholipids, to which ultrasonic vibrations and rotary evaporation were carried out to wipe off organic solvent until the gel formed. Ultrasonic buffer 1.5 mL was added and the compounds were rotated at room temperature after which 125 µL of calcium liquor was added to 0.5 mL of liposomes prepared through reverse-phase evaporation. After incubation at 37 ºC for 60 min, 12.5 µL of edetic acid (EDTA) was added to the compounds and shaken acutely, followed by incubation at 37 ºC for 15 min and at room temperature for another 30 min. Using an initial drug-to-lipid molar ratio of 5:1, the enveloped rate was 52%. After being stored at 4 ºC for 6 months, its permeability rate was less than 5%, which indicated the stability of 5-Fu-GCL was high. The recovery of 5-Fu-GCL was between 95.3% and 98.7%.
Pharmacokinetics of 5-Fu-GCL Two hundred and eighty-eight mice were randomly divided into four groups. Each group, which consisted of 36 males and 36 females, was injected with 5-Fu-GCL 20, 40, and 80 mg/kg or free 5-Fu 40 mg/kg through the tail vein. Before each injection and 2, 5, 10, 15, 20, 30, 45, 60, 90, 120, and 180 min later, 6 mice were anesthetized by diethyl ether, after which their eyeballs were excised and exsanguinated. Blood samples were centrifuged at 1400×g for 10 min to obtain 0.2 mL of serum each. Finally, mice were killed by dislocation of the cervical vertebra[9].
Tissue distribution of 5-Fu-GCL Twenty mice were stratified into two groups. Both groups including 5 males and 5 females were injected with 5-Fu-GCL 40 mg/kg and free 5-Fu 40 mg/kg through the caudal veins. Twenty minutes later, the mice were killed and the hearts, livers, spleens, lungs, kidneys, brains, lymph, thymuses, and marrow were removed, washed (except marrow), weighed, and homogenized using a tissue blender in 2 mL PBS solution. After extraction, concentrations in different tissues were detected by HPLC to study whether there were differences in distributions between the two groups[10].
Concentration-time course of 5-Fu-GCL in liver The livers from mice untreated and treated with 5-Fu-GCL 40 mg/kg or free 5-Fu 40 mg/kg at 2, 5, 10, 15, 20, 30, 45, 60, 90, 120, and 180 min were removed, washed, weighed, and homogenized using a tissue blender in 2 mL PBS solution. The sample was extracted and detected by HPLC.
Analytical method The drug concentration in serum and tissues were analyzed by HPLC. The mobile phase components were water and acetonitrile (99:1). Ultraviolet wavelength was 205 nm. The flow-rate was 1 mL/min[11,12].
The ratio of the peak area of 5-Fu to that of internal standard was plotted versus the concentration of the 5-Fu. Values of unknown serum drug concentrations were determined from calibration curve.
Standard solutions 5-Fu was dissolved in acetonitrile, then different amounts of this solution were added into blank 5-mL glass tubes and evaporated to dryness by nitrogen. The residues were dissolved with 0.2 mL mice serum or tissues to make 5-Fu of 0.313, 0.625, 1.25, 2.5, 5.0, 10.0, 20.0, and 40.0 mg/L, respectively, as serum standard solutions. The extraction of serum standard solutions was described in “Serum extraction procedure.”
Sample extraction procedure Serum or tissue samples (0.2 mL) were added with 150 µL para-amino-benzoic acid (15 mg/L) as internal standard, distilled water (0.2 mL), and ethyl acetate (4 mL). They were then vortex-mixed in a 10-mL glass tube for 1 min. After centrifugation at 700×g for 20 min, the organic phase was aspirated. The remainder was extracted by addition of 4 mL ethyl acetate as described above. The two organic phases were mixed and evaporated until dry under a gentle stream of nitrogen at 35 ºC in a water bath. The residues were reconstituted with 200 µL ethanol, and 50 µL of extracted samples were injected into the chromatography of liquid. On account of its character of being collapsed by ethyl acetate, 5-Fu-GCL could be detected as free 5-Fu. Both 5-Fu-GCL and free 5-Fu were detected by HPLC in the study.
Recovery, accuracy, and precision of 5-Fu Different 5-Fu standards were added into blank glass tubes and evaporated to dryness by nitrogen. The residues were dissolved with 0.2 mL serum or tissues to make concentrations of 0.313, 1.25, 5, and 20 mg/L. The samples were handled referring to the “serum extraction procedure.”
Five extracted samples of each concentration were determined. The recovery of 5-Fu in serum or tissues was assayed, and expressed as the percentage of detected concentrations to nominal concentration of standard (recovery). The accuracy of 5-Fu in serum or tissues was assayed, and expressed as the percentage of detected concentration to nominal concentration of serum or tissues (accuracy). Meanwhile, the inter-day and intra-day coefficient of variation of samples of different concentrations were measured 5 times on a single day and daily for a consecutive 5 d respectively (precision).
Pharmacokinetics calculations Phamacokinenic parameters were calculated by 3p97 program.
Statistical analysis Data were presented as mean±SD. Statistical analysis of the data was performed using the t-test. P<0.05 was considered statistically significant.
Results
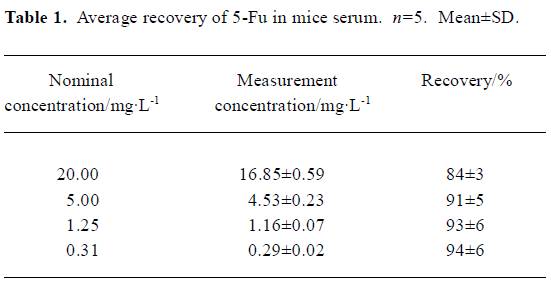
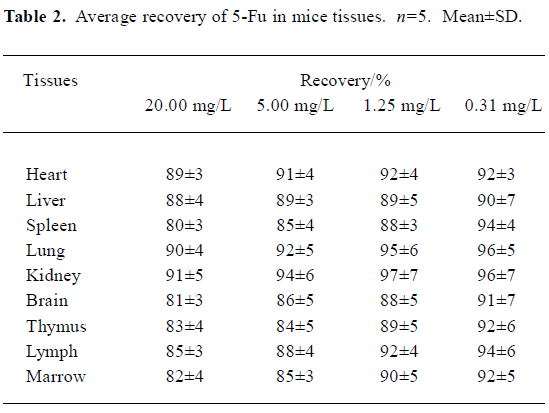
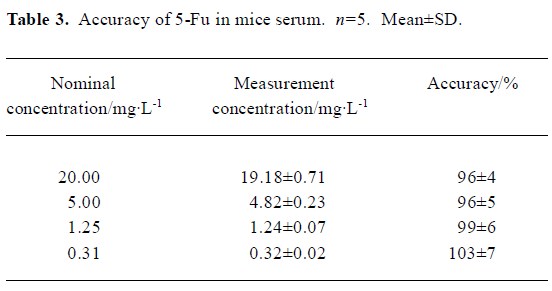
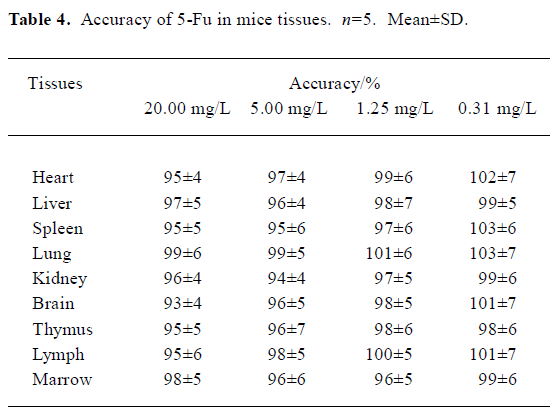
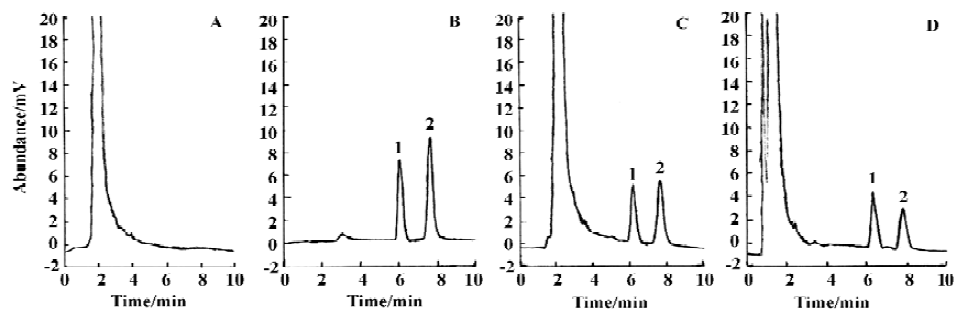
Method validation In the conditions described above, no significant interfering peak was observed. The retention time of 5-Fu was 7.7 min (Figure 1). The calibration curve of 5-fluorouracil in serum or tissue was in good linearity over the concentration range of -0.313–40.000 mg/L, and the coefficient of correlation was 0.9992–0.9996. The regression equation was: A=0.10+2.18C (serum); A=0.11+2.17C (heart); A=0.11+2.19C (liver); A=0.09+2.18C (spleen); A=0.13+2.19C (lung); A=0.10+2.17C (kidney); A=0.11+2.18C (brain); A=0.13+2.21C (thymus); A=0.12+2.20C (lymph); A=0.12+2.19C (marrow). A: ratio of 5-Fu peak area to internal standard peak area; C: 5-Fu concentration. The limit of detection was 15.6 ng. The average recovery of 5-Fu was over the range of (80±3)%–(97±7)% (Table 1, 2) and the accuracy of 5-Fu was over the range of (93±4)%–(103±7)% (Table 3, 4). The inter-day and intra-day reproducibility of 5-Fu is shown in Table 5, 6, and the coefficient of variation was all below 10%. Meanwhile, the average recovery of internal standard was over the range of (88±4)%–(93±5)%. And the stability of internal standard was high because the deviation of it were all below 15%.
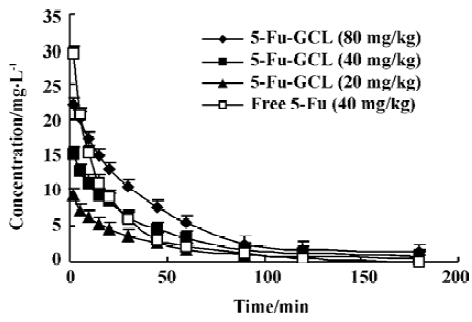
Pharmacokinetic parameters of 5-Fu-GCL in serum Data of drug serum concentrations at different time points in three 5-Fu-GCL groups (80, 40, and 20 mg/kg) and free 5-Fu group (40 mg/kg) are shown in Figure 2.
Pharmacokinetic parameters were calculated by 3p97 program. Drug serum concentration-time course of 5-Fu-GCL groups (80, 40, and 20 mg/kg) and free 5-Fu group (40 mg/kg) conformed to one compartment model of the first order absorption. Pharmacokinetic parameters of groups are shown in Table 7.
The results demonstrated that 5-Fu-GCL, in contrast to free 5-Fu, had delayed characteristics.
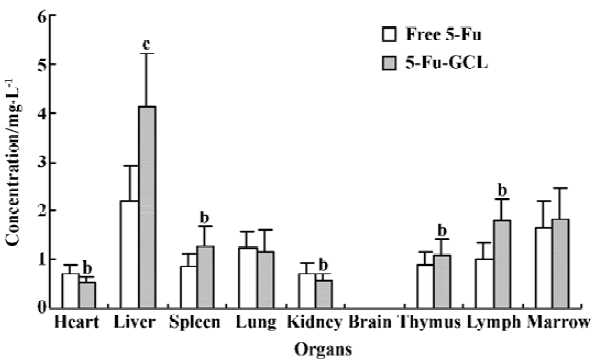
Concentration of 5-Fu-GCL in different tissue Distribution results showed that drug concentrations of 5-Fu-GCL in immune organs, which include the spleen, thymus, and lymph, but especially in the liver were remarkably higher than those of free 5-Fu at the same dose (40 mg/kg). Drug concentrations of 5-Fu-GCL in the heart and kidney were significant lower than that of free 5-Fu. In the brain, drug concentrations of both 5-Fu-GCL and free 5-Fu were not detected (Figure 3).
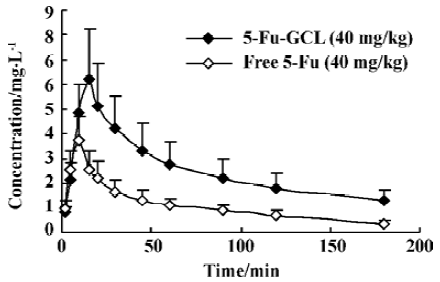
Concentration-time course of 5-Fu-GCL in liver The time course of drug concentration in the liver is shown in Figure 4. Area under time-concentration curve (AUC) of 5-Fu-GCL (485 mg·min·kg-1), which was calculated by 3p97 program, was three times higher than that of free 5-Fu (157 mg·min·kg-1).
Full table
Full table
Full table
Full table
Full table
Full table
Full table
Discussion
In drug delivery[13], liposomes could congregate drugs in certain tissues, decrease poisonous effects and increase the curative effect. Lecithoid material is the primary liposome in common use.
As GC is a cerebroside containing galactose, liposomes made of GC also possess galactose. As a result, this kind of liposome has a special bonding ability to salivary acid protein receptor located on the membrane of mammal[14,15]. Compared with lecithoid[16], GC has several advantages such as the stable chemical characters, the long retention in blood, the anti-oxide ability, and the unique directional trait. It could be developed to be a new kind of medicine carrier.
5-Fu is a dissoluble chemical with small molecules so it is difficult to achieve a highly enveloped rate. In a recent study, we applied combined calcium-induced fusion with reverse evaporation method to envelop 5-Fu, and the enveloped rate reached 52%.
Pharmacokinetic results showed that serum drug concentration curves of 5-Fu-GCL in single intravenous administration at 20, 40, and 80 mg/kg conformed to one compartment model of the first order absorption. Mean serum T1/2Ke of three dosage groups was similar. The pharmacokinetics of 5-Fu-GCL appeared to be linear over the doses. Both C0 and AUC were enhanced as the dose increased. T1/2Ke of 5-Fu-GCL was much longer and the C0 was strongly lower than those of free 5-Fu. All these results indicated that 5-Fu-GCL could be released much slower.
Because 5-Fu-GCL was injected through the vein and exhibited first order kinetic characteristics, it had no absorption or distribution phase and had only the elimination phase. For the study of tissue distribution one time point needed to be chosen. Tissue distribution results elucidated that the concentrations of 5-Fu-GCL in immune organs, especially in the liver, were remarkably higher than those of free 5-Fu with the same dose. Concentration-time course of 5-Fu-GCL in the liver showed that the AUC of 5-Fu-GCL was 3 times higher than that of free 5-Fu. This suggests that 5-Fu-GCL had a high selectivity to liver. In contrast, drug concentrations of 5-Fu-GCL in the heart and kidneys were significantly reduced, so we proposed that the toxicity of 5-Fu-GCL in the heart and kidneys were lower than that of free 5-Fu. Drug concentrations of both 5-Fu-GCL and free 5-Fu were not detected in the brain. The data indicate that they did not cross the blood brain barrier.
Previous studies showed that LD50 of 5-Fu-GCL was higher than that of free 5-Fu, indicating that toxicity of 5-Fu-GCL was lower than that of free 5-Fu. We, therefore conclude that 5-Fu-GCL could prospectively be used to develop a novel preparation for liver cancer therapy with long efficacy, low toxicity, and directional peculiarity.
Footnote
Project supported by Key Teacher Foundation of Ministry of Education of China (N
References
- Cao S, Rustum YM. Synergistic antitumor activity of irinotecan in combination with 5-fluorouracil in rats bearing advanced colorectal cancer: role of drug sequence and dose. Cancer Res 2000;60:3717-21.
- Di Paolo A, Danesi R, Falcone A, Cionini L, Vannozzi F, Masi G, et al. Relationship between 5-fluorouracil disposition, toxicity and dihydropyrimidine dehydrogenase activity in cancer patients. Ann Oncol 2001;12:1301-6.
- Schlemmer HP, Becker M, Bachert P, Dietz A, Rudat V, Vanselow B, et al. Alterations of intratumoral pharmacokinetics of 5-fluorouracil in head and neck carcinoma during simultaneous radiochemotherapy. Cancer Res 1999;59:2363-9.
- Fraile RJ, Baker LH, Buroker TR, Horwitz J, Vaitkevicius VK. Pharmacokinetics of 5-fluorouracil administered orally, by rapid intravenous and by slow infusion. Cancer Res 1980;40:2223-8.
- Hagiwara A, Sakakura C, Shirasu M, Yamasaki J, Togawa T, Takahashi T, et al. Therapeutic effects of 5-fluorouracil microspheres on peritoneal carcinomatosis induced by Colon 26 or B-16 melanoma in mice. Anticancer Drugs 1998;9:287-9.
- Jing M, Xi S, Chen R. The inhibitory effect of tissue plasminogen activator combined with 5-fluorouracil polyphase liposome on the scar formation in experimental filtration surgery. Chin J Ophthalmol 1997;33:376-80. Chinese.
- Wang G, Tucker IG, Roberts MS. In vitro and in vivo evaluation in rabbits of a controlled release 5-fluorouracil subconjunctival implants base on poly (D, L-lactide-co-glycoside). J Control Release 1996;13:1059-64.
- Umejima H, Kikuchi A, Kim NS, Uchida T., Goto S. Preparation and evaluation of Eudragit gels. VIII. Rectal absorption of 5-fluorouracil from Eudispert hv gels in rats. J Pharm Sci 1995;84:199-202.
- Kuan HY, Smith DE, Ensminger WD, Knol JA, De Remer SJ, Yang Z, et al. Regional pharmacokinetics of 5-fluorouracil in dogs: role of the liver, gastrointestinal tract, and lungs. Cancer Res 1998;58:1688-94.
- Zhao MZ, Zhao YK, Zhu LY, Xu G, Wang S, Mi JB, et al. Pharmacokinetics and tissue distribution of iv injection of polyphase liposome-encapsulated cisplatin (KM-1) in rats. Acta Pharmacol Sin 2003;24:89-92.
- Jung M, Berger G, Pohlen U, Pauser S, Reszka R, Buhr HJ. Simultaneous determination of 5-fluorouracil and its active metabolites in serum and tissue by HPLC. J Chromatogr B 1997;702:193-203.
- Joulia JM, Pinguet F, Grosse PY, Astre C, Bressolle F. Determination of 5-fluorouracil and its main metabolites in plasma by high-performance liquid chromatography: application to a pharmacokinetic study. J Chromatogr B 1997;692:427-35.
- Gregoriadis G. Engineering liposomes for drug delivery: progress and problems. Trends Biotechnol 1995;13:527-37.
- Nishikawa M, Hirabayashi H, Takakura Y, Hashida M. Design for cell-specific targeting of proteins utilizing sugar-recognition mechanism: effect of molecular weight of proteins on targeting efficiency. Pharm Res 1995;12:209-14.
- Murahashi N, Ishihara H, Sasaki A, Sakagami M, Hamana H. Hepatic accumulation of glutamic acid branched neogalactosyllipid modified liposomes. Biol Pharm Bull 1997;20:259-66.
- Tsuji H, Osaka S, Kiwada H. Targeting of liposomes surface-modified with glycyrrhizin to the liver. I. Preparation and biological disposition. Chem Pharm Bull 1991;39:1004-8.