
Effect of MePEG molecular weight and particle size on in vitro release of tumor necrosis factor-α-loaded nanoparticles1
Introduction
The rapid removal of colloidal drug carrier systems in blood by the mononuclear phagocytic system (MPS), which is comprised mainly of Kupffer cells of the liver and macrophages of the spleen, has been identified as major obstacle in the efficient targeting of colloidal particles in sites such as solid tumors and inflammatory regions[1,2]. Recently, a great deal of work has been devoted to developing “stealth” particles that are “invisible” to macrophages. Stealth nanoparticles (NP) are characterized by a prolonged half-life in the blood compartment that allows them to extravasate and accumulate more in pathological sites, such as tumors with leaky vasculature caused by the “enhanced permeability and retention effect”[3,4]. In our previous study, higher intratumoral drug accumulation and antitumor potency were achieved for recombinant human tumor necrosis factor-alpha (rHuTNF-α)-loaded stealth poly (methoxypolye-thyleneglycol cyanoacrylate-co-n-hexadecyl cyanoacrylate) (PEG-PHDCA) NP with sizes of approximately 150 nm and methoxypolyethyleneglycol (MePEG) molecular weight of 5000[5].
It is known that surface properties, particle size, and drug release behavior are the key for the biological fate of stealth NP. However, no previous study has focused on the relationship between these characteristics and their ability to accumulate in tumors and antitumor efficacy. The aim of the present study is to investigate the influence of MePEG molecular weight and particle size on their long-circulating properties, tumor targeting, and antitumor efficacy. In this article, rHuTNF-α-loaded PEG-PHDCA NP with various MePEG molecular weights and particle sizes were elaborated. The particle physicochemical characteristics, drug release behaviors and mechanisms in PBS and rat plasma were determined. This work on the in vitro characteristics of stealth NP might contribute to further understanding of their in vivo biological fate and tumor targeting properties.
Materials and methods
Reagents and drugs rHuTNF-α (Mr =17 000) was supplied by Shanghai Research Center of Biotechnology, Chinese Academy of Sciences (Shanghai, China). MePEG (Mr =2000, 5000), human serum albumin (HSA), and Na125I were obtained from Sigma Chemical (St Louis, MO, USA). MePEG (Mr =10 000) was donated by the Department of Polymer Material and Science of Fudan University. Cyanoacetic acid, 1,3-dicyclohexyl carbodiimide, 4-(dimethylamino) pyridine, and poly (vinylalcohol) (PVA) (Mr =16 000, 98% hydrolyzed) were purchased from Acros Organics (Geel, Belgium). All other reagents and solvents were of analytical grade.
Preparation and measurement of PEG-PHDCA polymers PEG-PHDCA was synthesized as described[6] in a previous study at an MePEG to n-hexadecyl ratio of 1:5 with appropriate modifications. The poly (n-hexadecyl cyanoacrylate) (PHDCA) was synthesized and used as a control.
1H-NMR and 13C-NMR spectra were recorded in CDCl3 on a Mercury Plus 400 MHz spectrometer. Recognition of methyl, methylene, methane and quaternary carbon nuclei in 13C-NMR spectra rested on the BB and DEPT-135. FTIR spectra (Avatar 360ESP) were obtained from a neat film cast from the chloroform copolymer solution between KBr tablets. GPC of polymers was performed in tetrahydrofuran with a Waters Associates Model ALC/GPC 244 apparatus. Molecular weight and molecular weight distribution of polymers were calculated by using polystyrene as the standard.
Preparation of rHuTNF-α-loaded nanoparticles rHuTNF-α was labeled with 125I using the IODO-GEN procedure. Briefly, 100 µg of protein in 60 µL of potassium phosphate buffer (0.1 mol/L, pH 7.4) was layered over a freshly prepared film of IODO-GEN (100 µg) and incubated at 4 ºC for 10 min with 2 mCi of carrier-free Na125I. The reaction mixture was made up to 0.5 mL volume with PBS, and the unreacted iodine was removed by gel filtration chromatography on a Sephadex G-25 PD10 column equilibrated with PBS. The specific radioactivity of the product was assessed in an autogamma (Packard Instruments, CT, Meriden, USA).
NP were prepared by an adjustment of the double emulsion (w1/o/w2) procedure as described in a previous study[7]. A w1/o emulsion was prepared by sonicating 0.1 mL of a rHuTNF-α solution (w1) containing HSA (2%, w/v) with a PEG-PHDCA or PHDCA solution in methylene chloride in an ice bath. The first emulsion was then poured into a PVA solution and sonicated in an ice bath. The double emulsion (w1/o/w2) obtained was diluted in 150 mL PVA solution (0.1%, w/v) and the organic solvent was evaporated at room temperature under reduced pressure. Finally, the NP were isolated and collected by two cycles of ultracentrifugation at 21 000×g for 40 min and washed three times with water.
Particle size and zeta potential measurement Particle size was determined in double distilled water by dynamic light scattering using Nicomp 380ZLS (NICOMP Particle Sizing Systems, Santa Barbara, CA, USA). Determinations were carried out at 23 ºC at a fixed angle of 90º. He-Ne laser was operated at 632.8 nm as light source.
Zeta potential of the NP was determined in double distilled water using electrophoretic light scattering technique on a Nicomp 380ZLS instrument.
Determination of entrapment efficiency and drug loading The NP containing rHuTNF-α and [125I]-iodinated rHuTNF-α with HSA (2.0%) were centrifuged, washed and the supernatants were assessed for gamma emission. The amount of rHuTNF-α encapsulated in the NPs was calculated by the difference between the total amount used to prepare the NP and the amount of rHuTNF-α present in the aqueous phase. The rHuTNF-α loading of the NP, given as a percentage, indicates the amount of protein encapsulated per 100 mg of NP.
rHuTNF-α release and MePEG liberation in PBS In vitro release studies were performed in triplicate in 25 mL of phosphate buffered saline (PBS, 0.05 mol/L, pH 7.4) containing 0.2% (w/v) sodium azide. The NP suspensions were continuously stirred in a thermoshake (60 r/min) at 37 ºC in dark. At predetermined time intervals, 1 mL samples were withdrawn, centrifuged at 21 000×g for 40 min, and replaced by fresh release medium. The 0.5 mL supernatant was removed for measurement of the released rHuTNF-α fraction, by radioactivity counting.
For PEG-PHDCA NP, MePEG liberated during drug release in PBS was assayed using an iodine complexation method[8]. At preselected times, 0.8 mL of supernatant was collected by centrifugation of NP suspensions at 21 000×g for 40 min, diluted with 1.2 mL double-distilled water and then mixed with 50 µL of iodine solution composed of I2 (10 g/L) and KI (20 g/L). After 5 min of incubation at room temperature in dark, the absorbance at 525 nm was read against a blank. In order to assay the total amount of MePEG in NP, the NP suspensions were digested by NaOH (2 mol/L) at 50 ºC for 5 d. The method was linear in the range of MePEG concentrations measured (1–25 mg/L).
rHuTNF-α release and nanoparticle degradation in rat plasma A total of 0.5 mL of each NP suspension was incubated with equal volumes of fresh rat plasma in a water bath at 37 ºC in dark. At 0.5, 1, 2, 3, 4, and 5 h, the NP were separated from release medium by centrifugation at 21 000×g for 40 min. The drug release (%) was determined at each time point from the ratio of radioactivity associated with the pellet and the supernatant[9].
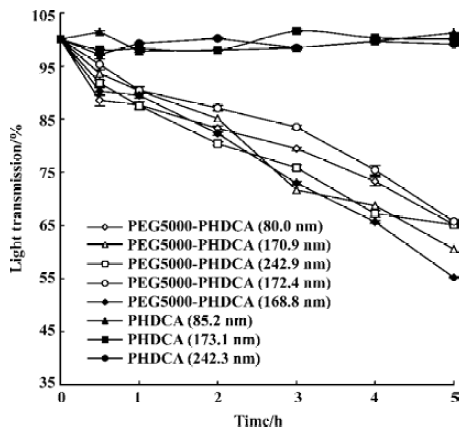
The degradation of the NP in rat plasma was performed by spectrophotometric measurements using a UV/VIS spectrophotometer (Shimadzu 2401) as described in a previous study[10]. The reduction in light transmission at 450 nm was recorded for evaluating the degradation of NP.
rHuTNF-α adsorption on PHDCA nanoparticles Blank PHDCA NP (approximately 25 mg of NPs in 25 mL of 0.05 mol/L phosphate buffer, pH 7.4, stabilized with 0.2% (w/v) sodium azide) were incubated at 37 ºC in a thermoshake (60 r/min) with an aqueous solution of rHuTNF-α (10 mg/L). At fixed time intervals, 1 mL of samples were centrifuged at 21 000×g for 40 min, and the amount of free rHuTNF-α in the supernatant was determined by radioactivity measurement. The amount of rHuTNF-α adsorbed on NP was determined using difference between the initial amount and the amount remaining in the supernatant.
Statistical analysis Physicochemical characteristics and in vitro release of NP were evaluated for statistical significance with analysis of variance (ANOVA) and Dunnett t-test.
Results
Characterization of PEG-PHDCA and PHDCA The 1H-NMR, 13C-NMR, and FTIR spectra were consistent with the structures of polymers (figures not shown). GPC showed that all polymers had narrow size distribution with an index of polydispersity less than 1.1 (Table 1).
Physicochemical characteristics of nanoparticles The basic characteristics of the eight types of NP formulations are summarized in Table 2.
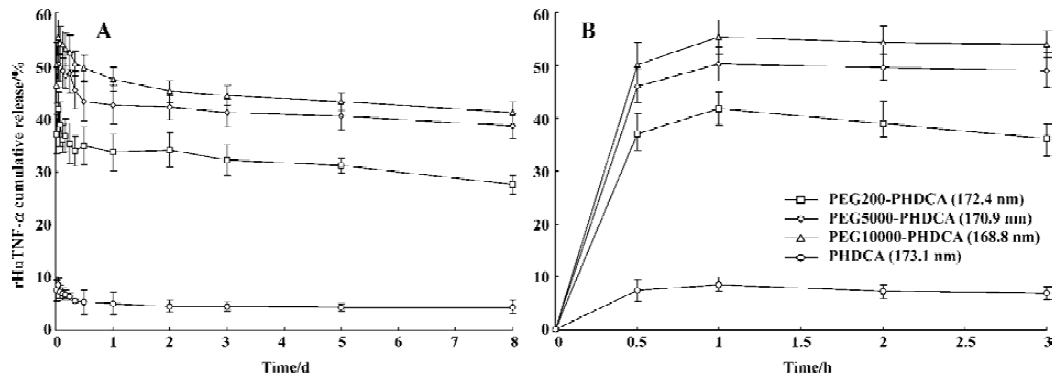
rHuTNF-α release and MePEG liberation in PBS The influence of MePEG molecular weight on the protein release in PBS is shown in Figure 1. For each formulation, an initial burst release followed by a slow reduction of rHuTNF-α cumulative release was observed. More than 40% of rHuTNF-α released from the PEG-PHDCA NP during the first 1 h, but less than 10% of the drug released from the PHDCA NP during the same time. At d 8, PEG-PHDCA NP had a remarkably higher cumulative release ratio than PHDCA (P<0.01). As a whole, the cumulative release ratio increased with the increase of MePEG molecular weight (P<0.05). The effect of particle size on the rHuTNF-α release is compared in Figure 2. Each formulation showed burst effect followed by decreased rHuTNF-α cumulative release. For both PEG5000-PHDCA and PHDCA NP, the NP of the smallest size showed the largest burst effect and highest cumulative release ratio (P<0.05).
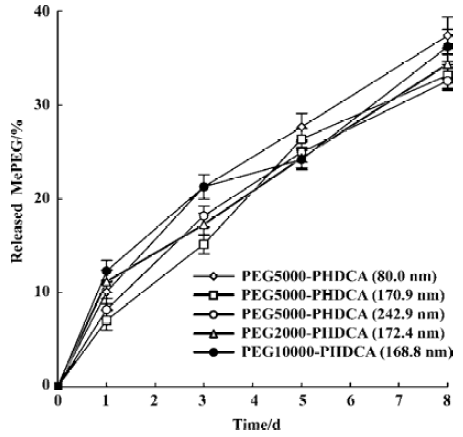
In the study of rHuTNF-α release in PBS, MePEG detachment from PEG-PHDCA NP was observed (Figure 3). In 8 d, about 35% (w/w) of MePEG liberated. The evolution of the mean diameters of the NP during the 8-d in vitro release period was studied. The initial size of all the NP formulations remained constant and no new larger or smaller size populations were observed untill the end of this study.
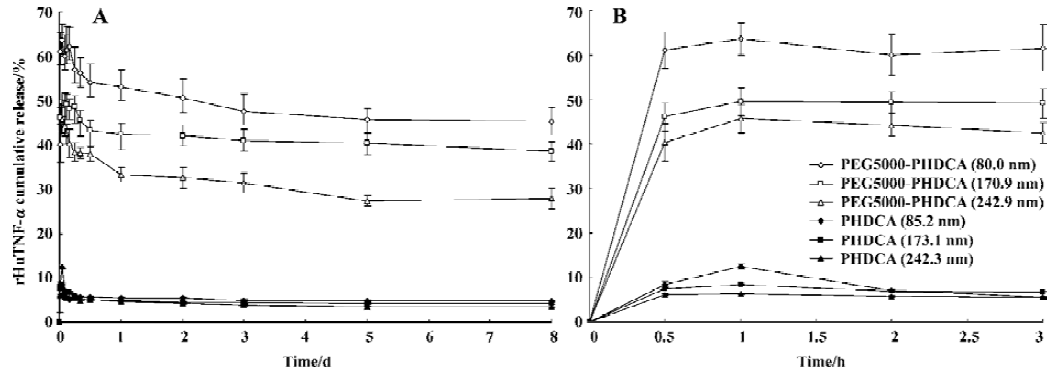
rHuTNF-α release and nanoparticle degradation in rat plasma All NP formulations showed a greater burst effect in rat plasma than in PBS (P<0.05) (Figure 4). The amount of drug released increased with the increase of incubation time. The burst release amount depended on both the particle size and the MePEG molecular weight. It significantly varied in the following series: PEG5000–PHDCA NP (80.0 nm)>PEG5000–PHDCA NP (170.9 nm)>PEG5000–PHDCA NP (242.9 nm) (P<0.01); PHDCA NP (85.2 nm)>PHDCA NP (173.1 nm)>PHDCA NP (242.3 nm) (P<0.01); PEG10000–PHDCA NP (168.8 nm)>PEG5000–PHDCA NP (170.9 nm)>PEG2000–PHDCA NP (172.4 nm) (P<0.01).
In rat plasma, more than 50% of PEG-PHDCA NP were degraded in 5 h of incubation, while no degradation occurred to PHDCA NP during the same time (Figure 5). For PEG-PHDCA NP, the degradation rate increased with the increase of MePEG molecular weight (P<0.01).
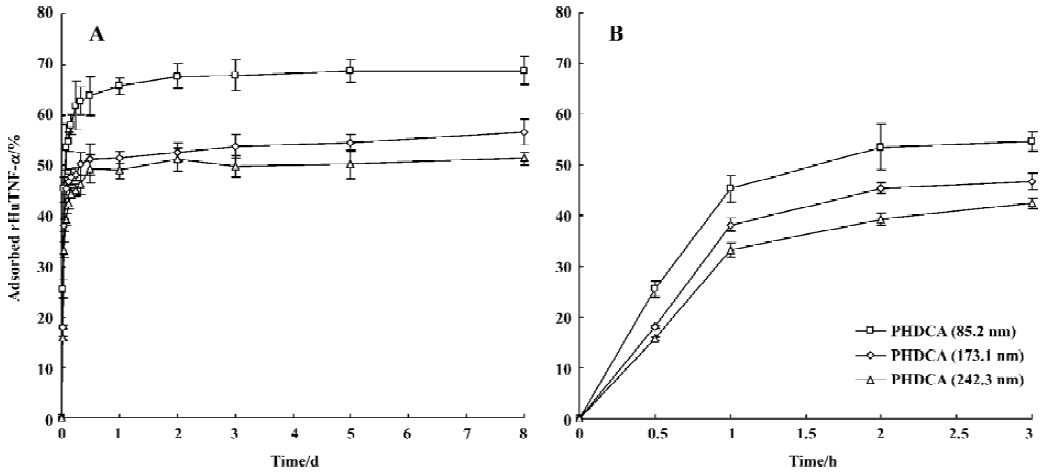
Evaluation of rHuTNF-α adsorption on PHDCA nanoparticles The aim of the present study was to shed light on the adsorption of rHuTNF-α on the hydrophobic blank PHDCA NP surface and to better understand what happens during its release from NP. The adsorption was studied for 8 d (Figure 6). The adsorption of rHuTNF-α on PHDCA NP was very rapid, depending on the particle size. It was shown that the adsorption capacity varies in the series: 85.2 nm>173.1 nm>242.3 nm (P<0.05). The adsorption plateau was reached after approximately 1 to 2 d.
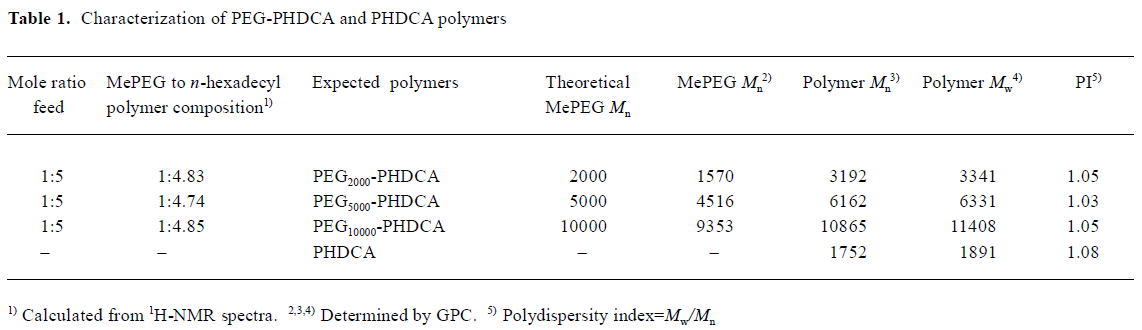
Full table

Full table

Discussion
Both particle size and MePEG coating influenced the zeta potential (Table 2). In the same size range, the significant lower negative potentials were obtained for PEG-PHDCA NP compared to PHDCA NP (P<0.01). This might be related to a shift of the hydrodynamic phase of shear to greater distance from the particle surface. The surface negative charge of PEG-PHDCA NP dramatically decreased with the increase of MePEG molecular weight. As for PEG5000-PHDCA NP, the surface charge notably increased when the particle size increased (P<0.05), and the reason is now under investigation.
In the same size range, PEG-PHDCA NP showed lower entrapment efficiency compared with PHDCA NP (P<0.01). The same results were obtained in the comparison made of PEG-PLA and PLA[11] and those made of PEG-PLGA and PLGA NP[12]. The difference might come from the presence of MePEG in the PHDCA chains, but the mechanism was indistinct. For PEG-PHDCA NP, entrapment efficiency increased with the increase of MePEG molecular weight (P<0.05), and the influence of particle size on the entrapment efficiency was irregular. No regular change with respect to drug loading was found. And the differences of drug loading among PHDCA and PEG-PHDCA NP were not remarkable (P>0.05).
The burst release depended on MePEG molecular weight (Figure 1). It was shown that the higher the MePEG molecular weight, the more the rHuTNF-α burst release. It was reported that the faster release of tetanus toxoid from PLA-PEG microspheres than PLA could be related to their different inner structure and reduced protein-polymer interactions[11]. Similarly, the MePEG chains in NP orient not only towards the external aqueous medium but also towards the inner aqueous phase[13,14]. Thus the protein reservoirs are theoretically surrounded by a MePEG barrier, which reduces the interaction of rHuTNF-α with the PHDCA matrix[7]. The different burst effect might result from the different intensity of this interaction between rHuTNF-α and PHDCA. It is supposed that the longest chain (MePEG of 10 000) hindered the interaction between rHuTNF-α and PHDCA, and PEG10000–PHDCA NP had the highest burst effect. Higher burst release of small size NP than middle and large NP could be related to their larger surface areas (Figure 2).
For PEG-PHDCA NP, a reduction of rHuTNF-α release percentage following burst effect was observed in PBS. This same phenomenon in the process of protein release from NP was reported in other articles[8,15]. MePEG chains can create a steric barrier around NP and repel proteins to some degree, in which the density of MePEG “brush” is of great importance[16]. MePEG chains are highly hydrophilic and can facilitate the penetration of incubation medium, and hydrolysis of the ester bonds associated to them[10] (Figure 3). After partial MePEG detachment, the density of MePEG “brush” was not sufficient to repel proteins and rHuTNF-α was reabsorbed on the particle surface, which resulted in a decreased cumulative release ratio. The readsorption phenomenon is evidenced in the study of rHuTNF-α adsorption on blank PHDCA NP (Figure 6).
There might be competition between protein release from NPs and protein readsorption onto the NP surface[15]. It is reasonable that the amount of rHuTNF-α detected in the supernatant was the result of rHuTNF-α release and rHuTNF-α readsorption onto the NP surface, each process having its own rate that depended on the rHuTNF-α concentrations in NP and in release medium. After a certain time, the release plateau was reached, indicating that equilibrium was established between the release and readsorption of rHuTNF-α. Noticeably, the readsorption phenomenon in the present study did not occur in a previous study in our lab[7]. We hypothesized that the loading of the NP used here was lower than that of the PEG5000-PHDCA NP (approximately 150 nm) prepared previously and relatively lower rHuTNF-α concentrations in NP were obtained in this study. Therefore it is possible that after a significant burst release, the rHuTNF-α concentration in NP is lower than that in the release medium and that a slow readsorption happened till the release plateau was reached. This could also be used to explain the readsorption of PHDCA NP.
The mean diameters of the eight types of NP remained constant during the 8-d release at 37 ºC in PBS. The ratios of particle size after incubation to its initial size for each NP formulation were all in the range of 1.0±0.3, indicating no significant aggregation or degradation occurred[17]. In summary, rHuTNF-α release was accompanied with constant particle size evolution and MePEG detachment from PEG-PHDCA NP in PBS. Combined with the known fact that polyalkylcyanoacrylate (PACA) polymer could be degraded mainly by enzymatic degradation[10], and PACA NP degraded mainly by surface erosion[18], it is hypothesized that rHuTNF-α release in PBS from PEG-PHDCA NP might follow a diffusion-based model.
Figure 5 showed that the presence of the MePEG chain greatly increased the degradation of PHDCA in rat plasma. This might be a result of the enhanced penetration of the plasma and the esterase hydrolysis caused by the highly hydrophilic MePEG moieties in the polymer structure.
The influence of particle size and MePEG molecular weight on the release of rHuTNF-α in rat plasma was similar to that in PBS. But no decreased cumulative release profiles was observed. We supposed that a competitive readsorption existed between rHuTNF-α and the proteins in rat plasma. Relatively small amounts of rHuTNF-α in the supernatant could hardly be reabsorbed onto the NP surface with a great amount of plasma proteins. Thus, a slightly increased cumulative release percentage was obtained under this condition. The burst release of PEG-PHDCA NP was greater than that of PHDCA NP. This could be a result of PEG-PHDCA degradation and the hindrance of interaction between protein and PHDCA by MePEG chains. Combined with the NP degradation behavior in rat plasma, it can be expected that rHuTNF-α released in rat plasma in a diffusion-controlled pattern for PHDCA NP, but in a diffusion and erosion-controlled manner for PEG-PHDCA NP.
Compared with PHDCA NP, PEG-PHDCA NP had a much higher cumulative release ratio, but their burst effects were a bit larger, which was a disadvantage when designing a stealth delivery system. However, compared to the free drug, a markedly extended circulating time was still obtained for stealth NP with high burst release[19]. And it was reported that in vitro release behavior did not always coincide with in vivo circulation time of NP, which represented the more complex nature of the in vivo system[20]. Therefore, for the validation of the “stealth” property of NP, pharmacokinetic studies in vivo are necessary. In view of the lower burst release possessed by PHDCA NPs, we supposed NP made with a proper ratio of PEG-PHDCA: PHDCA might have a lower burst effect as well as satisfactory cumulative release characteristics. This needs to be studied further.
In summary, a higher MePEG molecular weight offered higher entrapment efficiency, lower zeta potential, faster rHuTNF-α release rate and a larger cumulative release ratio. Larger particle size provided relatively higher surface charge, slower drug release rate and a smaller cumulative release ratio. rHuTNF-α release from PEG-PHDCA NP follows a diffusion-based model in PBS while a diffusion and erosion-controlled pattern in rat plasma. The present results implied that these NP might have different biological fates. The relationship between their in vitro characteristics and in vivo intratumoral drug accumulation and antitumor potency is now under investigation.
Acknowledgment
We thank Prof Jian-hua ZHU (Department of Radiopharmacy, Fudan University, Shanghai, China) for the radiolabelling and measurement.
Footnote
Project supported by the Science and Technology Development Foundation of Shanghai (N
References
- Dunn SE, Coombes AGA, Garnett MC, Davis SS, Davies MC, Illum L. In vitro cell interaction and in vivo biodistribution of poly (lactide-co-glycolide) nanospheres surface modified by poloxamer and poloxamine copolymers. J Control Release 1997;44:65-76.
- Mosqueira VCF, Legrand P, Morgat JL, Vert M, Mysiakine E, Gref R, et al. Biodistribution of long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm Res 2001;18:1411-19.
- Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev 2001;53:283-318.
- Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Del Rev 2002;54:631-51.
- Li YP, Pei YY, Zhou ZH, Zhang XY, Gu ZH, Ding J, et al. Stealth polycyanoacrylate nanoparticles as tumor necrosis factor-alpha carriers: pharmacokinetics and anti-tumor effects. Biol Pharm Bull 2001;24:662-5.
- Li YP, Zhou ZH, Pei YY, Zhang XY, Gu ZH, Yuan WF. PEGylated polycyanoacrylate nanoparticles as salvicine carriers: synthesis, preparation, and in vitro characterization. Acta Pharmacol Sin 2001;22:645-50.
- Li YP, Pei YY, Zhou ZH, Zhang XY, Gu ZH, Ding J, et al. PEGylated polycyanoacrylate nanoparticles as tumor necrosis factor-alpha carriers. J Control Release 2001;71:287-96.
- Zambaux MF, Bonneaux F, Gref R, Dellacherie E, Vigneron C. Protein C-loaded monomethoxypoly (ethylene oxide)-poly (lactic acid) nanoparticles. Int J Pharm 2001;212:1-9.
- Redhead HM, Davis SS, Illum L. Drug delivery in poly (lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: in vitro characterisation and in vivo evaluation. J Control Release 2001;70:353-63.
- Peracchia MT, Vauthier C, Desmaele D, Gulik A, Dedieu JC, Demoy M, et al. Pegylated nanoparticles from a novel methoxypolyethylene glycol cyanoacrylate-hexadecyl cyanoacrylate amphiphilic copolymer. Pharm Res 1998;15:550-6.
- Tobio M, Gref R, Sanchez A, Langer R, Alonso MJ. Stealth PLA-PEG nanoparticles as protein carriers for nasal administration. Pharm Res 1998;15:270-5.
- Li YP, Pei YY, Zhang XY, Gu ZH, Zhou ZH, Yuang WF, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis preparation and biodistribution in rats. J Control Release 2001;71:203-11.
- Brigger I, Chaminade P, Desmaele D, Peracchia MT, d’Angelo J, Gurny R, et al. Near infrared with principal component analysis as a novel analytical approach for nanoparticle technology. Pharm Res 2000;17:1124-32.
- Quellec P, Gref R, Perrin L, Dellacherie E, Sommer F, Verbavatz JM, et al. Protein encapsulation within polyethylene glycol-coated nanospheres. I. Physicochemical characterization. J Biomed Mater Res 1998;42:45-54.
- Quellec P, Gref R, Dellacherie E, Sommer F, Tran MD, Alonso MJ. Protein encapsulation within poly (ethylene glycol)-coated nanospheres. II. Controlled release properties. J Biomed Mater Res 1999;47:388-95.
- Passirani C, Barratt G, Devissaguet JP, Labarre D. Interactions of nanoparticles bearing heparin or dextran covalently bound to poly (methyl methacrylate) with the complement system. Life Sci 1998;62:775-85.
- Lin WJ, Juang LW, Lin CC. Stability and release performance of a series of pegylated copolymeric micelles. Pharm Res 2003;20:668-73.
- Muller RH, Lherm C, Herbort J, Couvreur P. In vitro model for the degradation of alkylcyanoacrylate nanoparticles. Biomaterials 1990;11:590-5.
- Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. PLGA-PEG nanoparticles of cisplatin: in vitro nanoparticles degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release 2002;79:123-35.
- Gaur U, Sahoo SK, De TK, Ghosh PC, Maitra A, Ghosh PK. Biodistribution of fluoresceinated dextran using novel nanoparticles evading reticuloendothelial system. Int J Pharm 2000;202:1-10.