
Effects of intrathecal 6-hydroxydopamine, α1 and α2 adrenergic receptor antagonists on antinociception of propofol in mice
Introduction
Propofol (2,6-diisopropylphenol, Diprivan) is widely used in the clinical setting as an iv anesthetic. Its antinociceptive effect results from an excitation of spinal GABAA receptors[1] and an inhibition of spinal NMDA, AMPA receptors[2], etc. A study by Antognini et al also suggested that propofol has a direct depressant effect on dorsal horn neuronal responses to noxious stimulation[3]. Therefore, these studies suggested a spinal mediation of propofol-induced antinociception. However there was also a study that concluded that propofol could control pain and this action might be centrally modulated through the opioid system rather than at the level of the spinal cord[4]. Therefore the contribution of supraspinal sites versus spinal sites to the antinociception of propofol is still unclear.
The mechanism whereby stimulation of opiate receptors in supraspinal regions produced analgesia has been extensively investigated and appeared, in part, to involve a descending noradrenergic pathway[5,6]. Norepinephrine (NE) is a major neurotransmitter in the descending inhibition of nociceptive transmission. Available evidence suggests that at the level of the spinal cord, there appear to be at least two neuronal systems that are involved. The first is the direct presynaptic inhibition of the nociception of the primary afferent neurons or postsynaptic inhibition of the second order neurons through activation of α2 adrenergic receptor (α2Rs)[7,8]. The second is the indirect activation of the inhibitory interneurons through α1Rs located on these neurons[8,9].
Based on these observations, in the present study, we injected propofol systemically, intrathecally, or intracere-broventricularly to further examine the role of spinal and supraspinal sites in mediating the antinociception of propofol. We sought evidences of the involvement of spinal NE, α1R, and α2R in the antinociception induced by sc propofol in mice.
Materials and methods
Animals With the approve of the Committee of Animal Research of Xuzhou Medical College, we conducted experiments on Kunming mice (22±3 g, Grade II, Certificate N
Chemicals Propofol and vehicle (10% intralipid) were supplied by the AstraZeneca Company. Prazosin, yohimbine and 6-hydroxydopamine (6-OHDA) were purchased from Sigma-Alexis (USA). Ice acetic acid was produced by Beijing Chemical Factory. All drugs were all freshly prepared. Prazosin was first dissolved in 50% dimethyl sulfoxide (Me2SO) in normal saline (NS) and the solution was diluted with NS. Yohimbine was dissolved in NS. 6-OHDA was dissolved in a saline vehicle (0.02% ascorbic acid in 0.9% saline). Systemic drug administration was sc performed in a single volume of 0.01 L/kg.
Intracerebroventricular injection in conscious mice[10] A 25-gauge needle was inserted in the left lateral ventricle of the brain with the following coordinates: left 2 mm of sagittal suture on the linking line of ears, and 2 mm down the skull surface. Volume of icv injections was 5 µL. The proper position of the icv localization was verified at the end of each protocol by injection of methylene blue staining with the same coordinates.
Intrathecal injection in conscious mice[2] A 25-gauge needle was inserted at L5–L6 intrathecal space. Ith placement was confirmed by a sudden lateral movement of the tail. All mice were injected with a 5-µL volume of drugs or vehicles and, after allowing 5 s for the injectant to disperse, the needle was slowly withdrawn. While developing this technique we injected 5 µL of 2% lidocaine ith in 10 mice, which consistently caused a transient hind-paw paralysis.
Tail-immersion test[11] The caudal 1/3 of tails of mice were immersed in 48±0.5 ºC water. The time for the mice to remove their tails from the water was expressed as the tail-flick latency (TFL). All mice were tested twice at a 5-min interval and the mean value was considered as the basal pain threshold. A cut-off time of 60 s was used to minimize damage to the skin of the tail. The influences of sc propofol (12.5, 25, and 50 mg/kg, n=10) alone on the TFL and the effects of pretreatment with ith prazosin and yohimbine 10 min before propofol (50 mg/kg, sc, n=10) injection on the TFL were investigated, respectively. To demonstrate the sc propofol-induced prolongation of TFL was not caused by its hypnotic effect, the time courses for the development of propofol-induced hypnosis were established. The loss of righting reflex was assessed by placing the mice on their backs and determining if the animals could right themselves within 30 s.
In a separate study, groups of mice were pretreated intrathecally with 6-OHDA (5, 10, and 20 µg) for 3 d (one injection in all). The dosing scheme of 6-OHDA was chosen according to the study by Hung et al[12] with minor modification. The tail-immersion test was performed on d 4 and the effect of propofol (50 mg/kg, sc, n=10) on TFL was observed.
Acetic acid-induced writhing test[2] Mice were administered NS, intralipid or propofol 30 min before ip injection of 0.9% ice acetic acid in a volume of 10 mL/kg. The number, latency and times of writhing mice were recorded within 15 min after the last injection.
Statistical analysis All data were present as mean±SD. Statistical comparisons in each group were performed by one-way analysis of variance (ANOVA) followed by t-test. Difference was considered to be significant at P<0.05.
Results
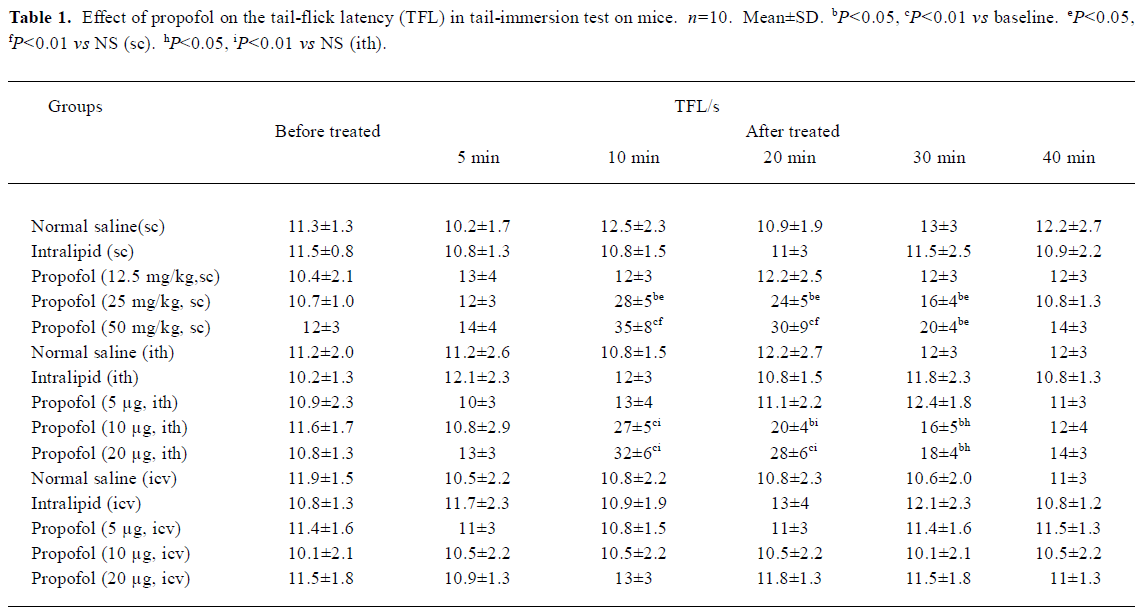
Hypnotic and antinociceptive effects of propofol were temporarily uncoupled Propofol 12.5, 25 mg/kg sc had no appreciable effect on the general behavior in mice. After 2–5 min treatment of propofol (50 mg/kg, sc), mice showed slight sedation. The sedative effect reached maximum after 5 min of propofol injection, and 20% mice (2/10) developed a loss of righting reflex; however, after this time there was no significant increase in TFL. In contrast, sc propofol resulted in a significant increase in TFL 10–30 min after its injection with the peak effect at 10 min (P<0.01, Table 1), but after 30 min, all mice remained righting reflex and there was no appreciable effect on general behavior. Neither propofol ith nor icv resulted in any appreciable abnormal behavior.
Effects of propofol on pain threshold in tail-immersion test and acetic acid-induced writhing test in mice There was no significant change of TFL before and after sc, ith, or icv injection of 10% intralipid compared with the NS (sc, ith, or icv) group, respectively in tail-immersion test (P>0.05, Table 1). Propofol (12.5 mg/kg, sc) and propofol (5 µg, ith) showed no effect on TFL. However, propofol (25 and 50 mg/kg, sc) and propofol (10, 20 µg, ith) increased the TFL in a dose- and time-dependent manner 10–30 min after its injection(P<0.05 or P<0.01 vs NS, Table 1).
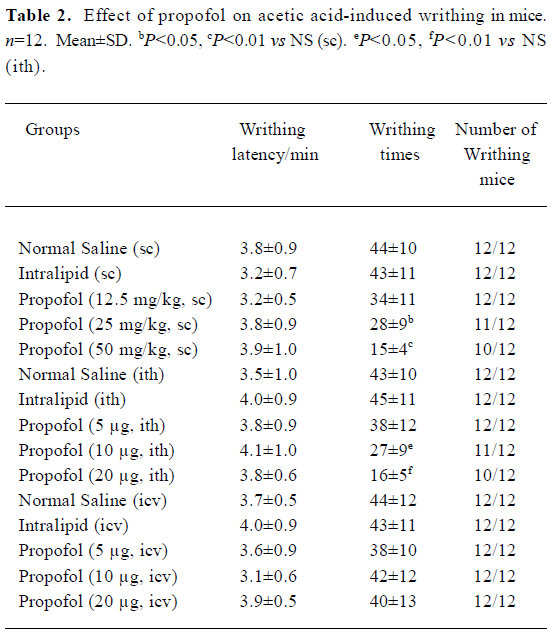
No significant difference existed in the number of writhing mice, writhing latency writhing times before and after sc, ith, or icv injection of 10% intralipid. Propofol (12.5 mg/kg, sc) and propofol (5 µg, ith) showed no anti-nociceptive effect in acetic acid-induced writhing test. But propofol (25 and 50 mg/kg, sc) and propofol (10 and 20 µg, ith) significant reduced the writhing times in a dose-dependent way(sc or ith) group (P<0.05 or P<0.01 vs NS, Table 2). Icv propofol (10, 20, and 40 µg) did not produce any antinocicep-tive effects in the tail-immersion test and acetic acid-induced writhing test (P>0.05, Table 1, 2).
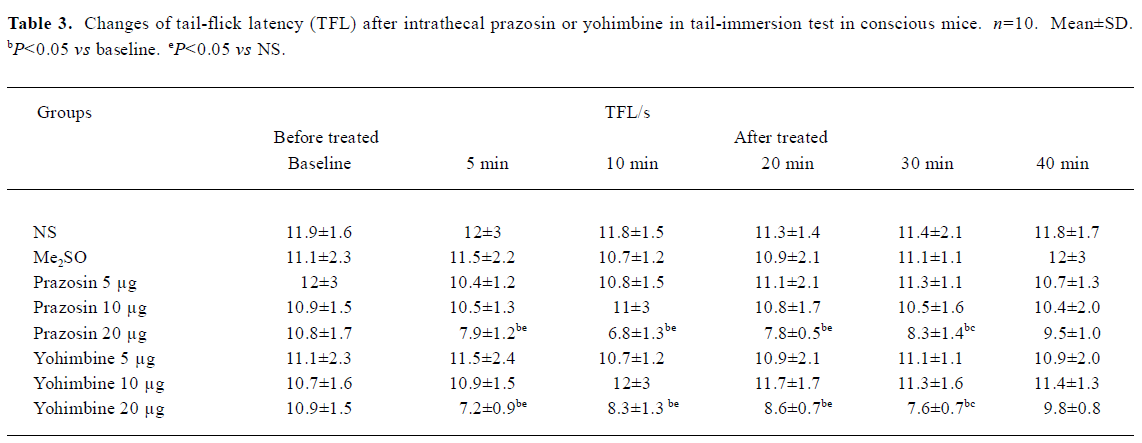
Effects of intrathecal prazosin and yohimbine on TFL in conscious mice in tail-immersion test No significant difference existed in the TFL between NS group and Me2SO group (P>0.05, Table 3). Ith prazosin (5 and 10 µg) or yohimbine (5 and 10 µg) alone had no effect on the general behavior and TFL. However, mice in prazosin (20 µg) and yohimbine (20 µg) groups exhibited less movement and the muscle strength of hindlimbs decreased at least for 1 h followed by a full recovery. The TFL in prazosin 20 µg and yohimbine 20 µg groups was significantly shortened, compared with its own baseline and NS group (P<0.05, Table 3). Therefore, prazosin (5 and 10 µg) and yohimbine (5 and 10 µg) were used in the subsequent antinociceptive studies of propofol.
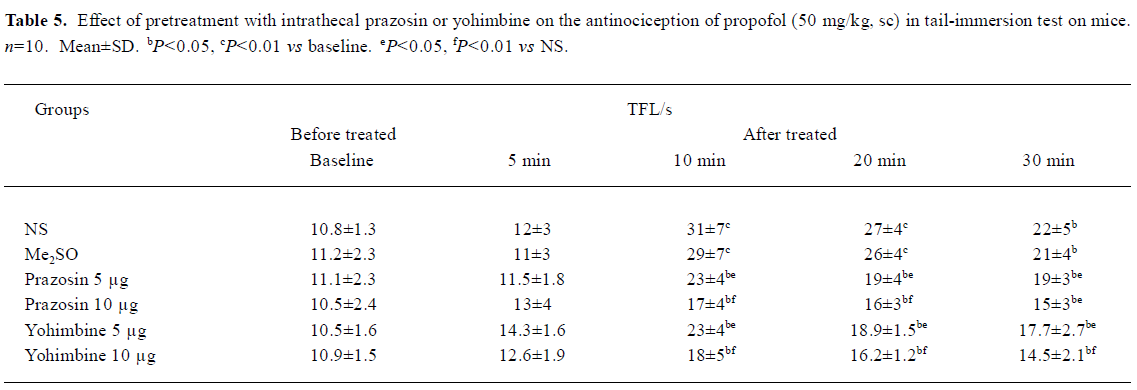
Effects of pretreatment with intrathecal prazosin and yohimbine on TFL in propofol (50 mg/kg, sc)-treated mice in tail-immersion test The mice were injected intrathecally with NS, Me2SO, prazosin, or yohimbine at different doses 10 min before sc propofol. No significant differences existed in the TFL between NS group and Me2SO group. Pretreatment with ith prazosin (5, 10 µg) and yohimbine (5, 10 µg) significantly decreased the TFL 10-30 min after propofol injection with the doses of prazosin and yohimbine increasing (P<0.05 vs NS, Table 5).
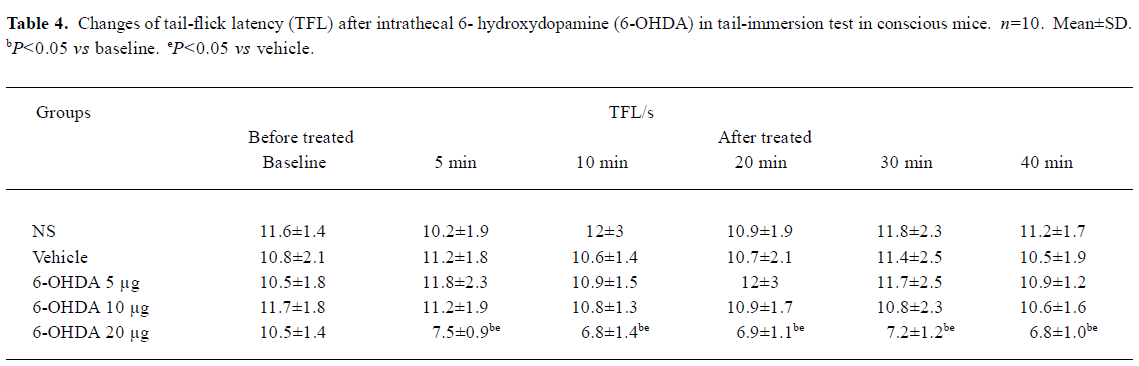
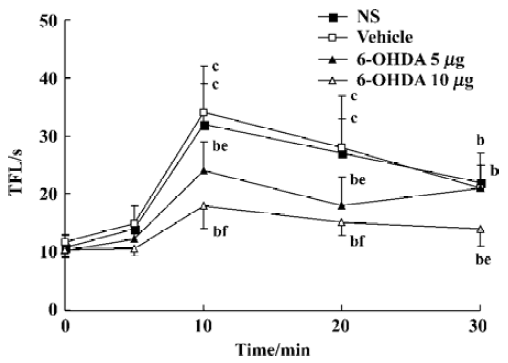
Effects of pretreatment with 6-OHDA on TFL in conscious mice and propofol (50 mg/kg, sc)-treated mice in tail-immersion test Mice in the 6-OHDA (20 µg) alone group exhibited biting and scratching behavior and the TFL significantly decreased in comparison with its own baseline and the NS group (P<0.05, Table 4). No change in behavior and TFL was observed after treatment with ith NS, vehicle or 6-OHDA (5, 10 µg) in conscious mice. However, pretreatment with ith 6-OHDA (5, 10 µg) significantly decreased the TFL 10–30 min after propofol treatment with the dose of 6-OHDA increasing (P<0.05 vs NS, Figure 1).
Full table
Full table
Full table
Full table
Full table
Discussion
In attempting to eluciate the mechanisms for the antinociceptive effect of propofol, it is important to obviate sedation that can exert on the antinociceptive assay. In the present study, we found that the hypnotic and antinociceptive effects of propofol were temporarily uncoupled. In addition, the anesthetic on awareness was clearly supraspinal. Ith 6-OHDA, prazosin, or yohimbine significantly inhibited the antinociception of propofol but not consciousness of animals. Furthermore, we used the heat tail-flick assay, which measures the latency of a spinal withdraw reflex to noxious heat, which is independent of hypnotic-induced decrement in purposeful movement. These results indicated that propofol-induced prolongation of TFL observed in the present study was a result of antinociception but not sedation.
Propofol (50 mg/kg, sc) produced a motor block 2–5 min after injection, but there was no significant increase in TFL. Furthermore, Kerz T et al have suggested that immobility during propofol anesthesia was not caused by a depression of spinal motoneuron circuit excitability[13]. In addition, pretreatment with ith 6-OHDA, prazosin, or yohimbine inhibited the antinociception of propofol but not propofol-induced motor impairment. Therefore, it is not probable that motor nerve conduction block affects TFL nonspecifically.
In the present study, propofol (25 and 50 mg/kg, sc) caused no noticeable sedation 30 min after its injection, but dose-dependently decreased the writhing times in the acetic-acid writhing test on mice. Therefore, these results suggested that systemic propofol also inhibited the chemical stimulation. The same results were shown in a previous study[2]. However, propofol had no effect on the writhing latency, which suggested that the antinociceptive effect of propofol at subanesthesia doses on inflammatory pain might be not very strong.
In the present study, systemic propofol (25 and 50 mg/kg, sc) and propofol (10 and 20 µg, ith) displayed a dose- and time-dependent antinociceptive effect. However, icv propofol did not produce any antinociceptive effects. Taken together, these results suggest that propofol produced antinociception at the spinal level but not at the supraspinal sites.
Ith 6-OHDA (20 µg) treatment for 3 d, which markedly depleted NE contents by more than 90% but not serotonin in the spinal cord study[12], significantly shortened the basal TFL in conscious mice in our study. Additionally, α1R antagonist prazosin (20 µg, ith) or α2R antagonist yohimbine (20 µg, ith) alone significantly shortened the basal TFL. These results supported the previous finding that noradrenergic synaptic transmission was important in pain sensory processing in the spinal dorsal horn. The effect of ith high dose of yohimbine on tail response was consistent with the conclusion that NE mediated the inhibition of nociception by α2Rs in the spinal cord. The reduction of TFL after ith higher dose of prazosin suggested that spinal α1Rs also contributed to the pain sensory modulation, which agrees with the purpose that NE mediated the inhibition of nociception by activation of α1Rs located on inhibitory interneurons[8,9]. This possibility is supported by several studies which demonstrated that ith α1R agonist activated dorsal horn to produce antinociception. For example ith selective α1R agonists, such as methoxamine[14,15] and phenylephrine[16] produced antinociception that could be reduced by pretreatment with ith α1R antagonist prazosin[14,15].
Pretreatment with ith 6-OHDA (5 and 10 µg), which markedly depleted NE contents in the spinal cord, significantly attenuated the antinociception of propofol. Furthermore, pretreatment with ith α1R antagonist prazosin or α2 R antagonist yohimbine significantly inhibited the antinociception of sc propofol in tail-immersion test. Therefore, these results strongly indicated that spinal noradrenergic system had a significant modulatory role in propofol antinociception. However, ith 6-OHDA, prazosin and yohimbine did not completely reverse the antinociception of propofol. This suggested that other mechanisms, such as an excitation of spinal GABAA receptors[1] and an inhibition of spinal NMDA, AMPA receptors[2], contributed to antinociception of propofol. In the present study, we used only α1R and α2R antagonists; therefore it was unclear whether propofol could bind directly with α1Rs or α2Rs in the spinal cord.
Taken together, our data suggest that systemic propofol activates the central noradrenergic system to produce antinociception, which increases the spinal release of NE and results in an antinociceptive effect via α1Rs and α2Rs in the spinal cord. To further prove this speculation, a direct measurement of the spinal release of NE induced by propofol will be necessary in the near future.
References
- Nadeson R, Goodchid CS. Antinociceptive properties of propofol: involvement of spinal cord gamma-aminobutyric acid (A) receptors. J Pharmacol Exp Ther 1997;282:1181-6.
- Xu AJ, Duan SM, Zeng YM. Effects of intrathecal NMDA and AMPA receptors agonists or antagonists on antinociception of propofol. Acta Pharmacol Sin 2004;25:9-14.
- Antognini JF, Wang XW, Piercy M, Carstens E. Propofol directly depresses lumbar dorsal horn neuronal responses to noxious stimulation in goats. Can J Anaesth 2000;47:273-9.
- Anwar MM, Abdel-Rahman MS. Effect of propofol on perception of pain in mice: mechanisms of action. Comp Biochem Physiol A Mol Integr Physiol 1998;120:249-53.
- Fang F, Proudfit HK. Antinociception produced by microinjection of morphine in the rat periaqueductal gray is enhanced in the foot, but not the tail, by intrathecal injection of alpha1-adrenoceptor antagonists. Brain Res 1998;790:14-24.
- Feng CY, McAdoo DJ, Willis WD. Periaqueductal gray stimulation-induced inhibition of nociceptive dorsal horn neurons in rats is associated with the release of norepinephrine, serotonin, and amino acids. J Pharmacol Exp Ther 1999;289:868-76.
- Pan YZ, Li DP, Pan HL. Inhibition of glutamatergic synaptic input to spinal Lamina IIo neurons by presynaptic α2-adrenergic receptors. J Neurophysiol 2002;87:1938-47.
- Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology 2000;92:473-84.
- Baba H, Goldstein PA, Okamoto M, Kohno T, Ataka T, Yoshimura M, et al. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 2): effects on somatodendritic sites of GABAergic neurons. Anesthesiology 2000;92:485-92.
- Zhang CY, Zhu TJ, Hu GX, Chen XY, Liu DX, Chen ZK. Effect of sodium dimercaptopropanesulfonate on antagonism of tetramethyl-enedisulphotetramine to GABA receptor. Acta Pharmacol Sin 2001;22:435-9.
- Xu SY, Bian RL, Chen X. Methodology of pharmacological experiments. 3 rd ed. Beijing: People’s Medical Publishing House; 2002. p 882–7.
- Hung KC, Wu HE, Mizoguchi H, Leitermann R, Tseng LF. Intrathecal treatment with 6-hydroxydopamine or 5,7-dihydroxytryptamine blocks the antinociception induced by endomorphin-1 and endomorphin-2 given intracerebroventricularly in the mouse. J Pharmacol Sci 2003;93:299-306.
- Kerz T, Hennes HJ, Feve A, Decq P, Filipetti P, Duvaldestin P. Effects of propofol on H-reflex in humans. Anesthesiology 2001;94:32-7.
- Loomis CW, Arunachalam KD. Intrathecal St-587: effects on nociceptive reflexes and blood pressure in the rat. J Pharmacol Exp Ther 1992;263:428-35.
- Howe JR, Wang JY, Yaksh TL. Selective antagonism of the antinociceptive effect of intrathecally applied alpha adrenergic agonists by intrathecal prazosin and intrathecal yohimbine. J Pharmacol Exp Ther 1983;224:552-8.
- Aran S, Proudfit HK. Antinociceptive interactions between intrathecally administered alpha noradrenergic agonists and 5'-N-ethylcarboxamide adenosine. Brain Res 1990;519:287-93.