
Effects of adenosine agonist R-phenylisopropyl-adenosine on halothane anesthesia and antinociception in rats1
Introduction
Adenosine, a purine nucleotide, is present in the brain in concentrations sufficient to be important in the regulation of central nervous system (CNS) function[1]. The compound adenosine has various modulatory effects in the peripheral and central nervous system, mediated through specific cell-surface associated receptors[2]. For example, in addition to the antinociceptive action of adenosine in animal models[3], adenosine is used to treat the symptoms of paroxysmal supraventricular tachycardias, and to produce controlled hypotension during some surgical procedures, and in the diagnosis of coronary artery disease[4]. The current view is that adenosine receptors of the A1 subtype are associated with a modulatory effect on pain transmission at brain and spinal cord level. Animal studies have repeatedly demonstrated that adenosine-mediated inhibitory influences presumed nociceptive reflex responses[5]. These examinations on rodents have tested acute pain models involving tactile, pressure and heat stimulations. More recently, animal lesion models, presumably reflecting chronic pain, have shown that adenosine analogue can suppress nociceptive behavior both by systemic and intrathecal administration[6]. Consequently, there are substantial evidence that adenosine can modulate nociceptive input. The general thinking about the mechanism of analgesic-anesthetic action of adenosine is that adenosine interacts with G-protein-coupled adenosine receptor and activates K+ channel in CNS. However the roles of K+ channel activation by adenosine in the antinociceptive action of adenosine in CNS and spinal cord has not been well demonstrated. Thus, in the present study we investigated whether adenosine agonist R-phenylisopropyl-adenosine (R-PIA) administered through intracere-broventricular (ICV) or intrathecal (IT) methods will produce an antinociceptive effect and enhance the halothane-anesthetized state.
Materials and methods
Animal preparation With approval of the Animal Care and Use Committee of our institution, studies were performed on 70 male Sprague-Dawley rats weighing 300±20 g (9 weeks old). Rats were housed individually in a temperature-controlled (21±1 ºC) room with a 12-h light/dark cycle, and they were given free access to water and food. All experimental measurements were performed between 10:00 AM and 5:00 PM. Each rat was assigned to only one of the following protocols: minimum alveolar concentration (MAC) of halothane (n=30), or tail-flick latency (IT injection, n=20; ICV injection, n=20). Each animal was studied two to four times in an experimental series with an interval of at least 5 d.
Surgical preparation For the ICV cannular placement, as previously described by Ma et al[7], animals were anesthetized with pentobarbital (50 mg/kg), and positioned in a stereotaxic apparatus (Narishige, Tokyo, Japan). A 24-gauge stainless steel guide cannula was unilaterally implanted 1 mm above the lateral ventricle using the following stereotaxic coordinates: 1.5 mm lateral to the midline, 1.0 mm posterior to bregma, 2.5 mm ventral to dura. The guide cannula was then fixed to the skull with two steel screws and dental cement. The IT catheter was implanted as previously described by Zeng et al[8], and animals were placed in a stereotaxic head holder with the head fixed forward. At the site of occipital a midline incision was made till the escape of cerebrospinal fluid. Intrathecal catheter was inserted at a length of 8.5 cm to the lumbar intrathecal level. The catheter’s external arm was tunneled subcutaneously to emerge at the neck. After the surgery the rats were allowed to recover for one week before the experiments began. Only animals exhibiting no motor deficits as a result of surgery were used.
Minimum alveolar concentration measurement Anesthesia was induced through inhalation of halothane in a transparent container. The rat’s trachea was intubated with a 16-gauge cannula, and the lungs were mechanically ventilated with 1.0% halothane in oxygen and air (FIO2 0.3–0.5). End-tidal carbon dioxide pressure was maintained at 35 to 40 mmHg. Rectal temperature was continuously monitored and maintained at 37.5 ºC with a heating pad. Fifteen minutes after the initiation of halothane anesthesia, saline or 4-aminopyridine (4-AP) 2 mg/kg was injected intraperitoneally, then a 30-gauge stainless steel internal cannula connected to polyethylene tubing was inserted into the guide cannula and positioned 1.0 mm beyond the tip. R-PIA at the dose of 0.8 µg, 1.0 µg, and 2.0 µg, or combined with A1 antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) 5.0 µg was injected into the lateral cerebral ventricle at a volume of 5.0 µL over 90 s using a microinjection pump. Five minutes after the drug or vehicle injection, MAC was determined using the up and down technique[9]. Briefly, the administration of halothane was adjusted in steps of 0.1%, and a stable end-tidal concentration for 15 min was obtained before stimulation. Noxious stimulation was applied with a 6-inch hemostat to the middle third of the tail for 60 s at the first ratchet position. The criteria for positive movements included purposeful movements of either the head or the four extremities. When animal had a positive response, the halothane concentration was increased; when there was no response, the concentration was decreased until movement was observed. When the interval was bracketed by positive and negative responses, the midpoint of the interval was then the MAC of halothane. End-tidal gas samples were obtained with an airtight glass syringe through a 26-gauge needle inserted to a tracheal tube during 15 expirations. Halothane concentrations were analyzed using an infrared analyzer (M1025B; Hewlett Packard). Calibration with the standard gas was performed before study. All chemicals were purchased from Sigma Chemical (St Louis, MO, USA).
Measurements of antinociception The antinociceptive effect was measured by the tail-flick (TF) latency response. A high-intensity light was focused on the dorsal surface of the rat tail; the time for the rat to move its tail out of the light beam was automatically recorded (Thermal Analgesimeter KN-205E, Natume, Tokyo, Japan). A different patch of the middle one-third portion of the tail was exposed to the light beam on each trial to minimize the risk of tissue damage during the experiment. A cut-off time of 10 s was predeter-mined, at which time the trial was terminated if no response occurred. TF latency was determined 5, 10, 15, 20, 30, 40, 50, and 60 min after ICV or IT administration of R-PIA (0.8 µg, 1.0 µg, and 2.0 µg), with or without pretreatment of DPCPX 5.0 µg or 4-AP 2 mg/kg. Each TF latency data point consisted of a mean of three trials on an individual animal.
Histology examination At the end of the experiments, bromophenol blue (5.0 µL) was microinjected to label the site of ICV injection. The rat was then killed with an overdose of pentobarbital. The brain was removed and fixed in 10% neutral buffered formalin. Dye spots were localized from 0.3–0.5 mm serial coronal sections and identified on diagrams from the atlas of Paxinos and Watson[10]. Bromophenol blue (10 µL) was used to confirm the position of the intrathecal catheter and likely spread of the injectate.
Statistical analysis All data were presented as mean± SEM. In MAC measurements statistical testing were performed with a Student’s t-test. Because a cut-off value was used in the TF latency test, data were converted to the percentages of the maximum possible effect (% MPE). Where % MPE=(postdrug TF latency-baseline TF latency)/(cut-off time-baseline TF latency)×100. The cut-off time was defined as a stimulus time of 10 s. For the effect of drugs on TF latency, statistical differences were analyzed using a two-way analysis of variance (ANOVA), followed by Fisher’s test for post hoc analysis of means. P<0.05 was considered to be statistically significant.
Results
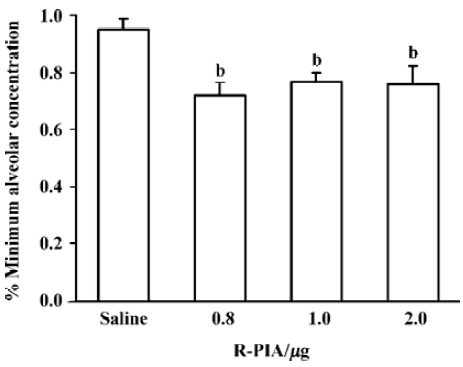
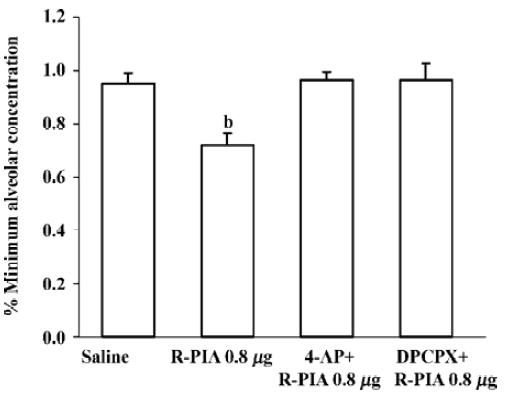
Effect of ICV R-PIA injection on the MAC of halothane with pretreatment 4-AP or DPCPX The MAC of halothane was significantly reduced by direct application of adenosine agonist R-PIA in doses of 0.8 µg, 1.0 µg, and 2.0 µg. The MAC of halothane in the control group (saline injection) was 0.95%±0.05% (Figure 1), which corresponds with our previously reported MAC values[9]. R-PIA 0.8 µg decreased MAC of halothane by 29%. Pretreatment of 4-AP 2 mg/kg intraperitoneal injection or ICV injection DPCPX 5 µg inhibited the effect of R-PIA 0.8 µg, whereas the dose of 4-AP or DPCPX itself did not affect the MAC of halothane (Figure 2).
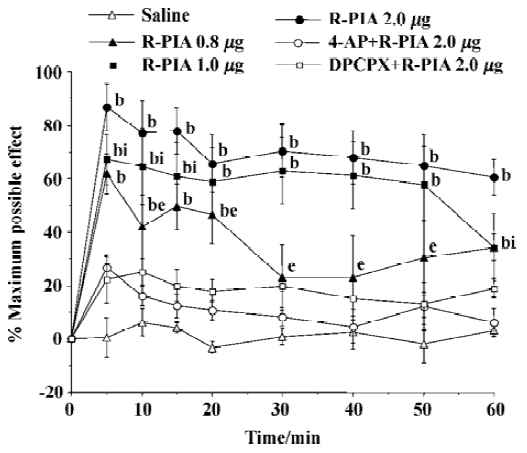
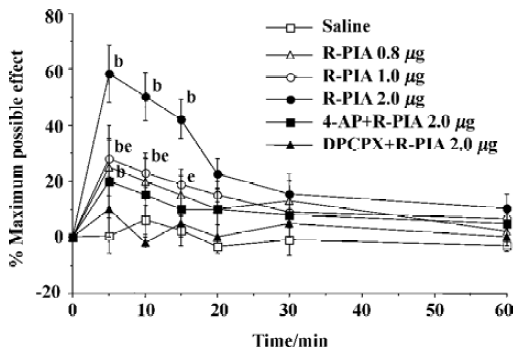
ICV and IT administration of R-PIA on antinocicep-tion Time courses were determined for adenosine agonists R-PIA in the TF latency test. IT administration of R-PIA in doses of 0.8–2.0 µg induced antinociception as reflected by the increase in latency times compared with the control animals. The antinociceptive action reached maximal effect within 10 min and lasted over 60 min, and showed a dose- dependent manner (Figure 3). The ICV administration of adenosine agonist R-PIA (0.8–2.0 µg) produced the antinociceptive effect as reflected by increasing the TF latency (Figure 4). The peak effect of antinociception of R-PIA was within 5 min and showed a dose-dependent manner. The intraperitoneal injection of 4-AP or ICV injection of DPCPX had no effect on the baseline of TF latency, but its pretreatment produced a significant reverse effect elicited by the ICV or IT administration of R-PIA (Figure 3, 4). The experiment dose of R-PIA did not show any detectable effect on motor function or general behavior during the observation period (60 min).
Discussion
The present data demonstrated that adenosine agonist R-PIA injection by ICV and IT, produced antinociceptive effect in a dose-related manner and enhanced halothane anesthesia. Because such effects of R-PIA were attenuated with pretreatment of DPCPX, an A1 receptor antagonist, and 4-AP, a voltage-gated K+ channel blocker, the action of R-PIA is mediated via A1 receptor subtype activation and its mechanisms are likely to be involved in, at least in part, K+ channel activation.
Adenosine and its analogues were shown to produce antinociception in the tail flick and hot plate tests following both IT and central administration[5,6]. Such antinociception has been diminished by methylxanthines, such as caffeine and theophylline, suggesting the involvement of specific adenosine receptor[11]. In the present study, we administered R-PIA through IT method to induce dose-dependent antinociception. Pretreatment A1 antagonist DPCPX reversed this effect. The results indicate that R-PIA inhibits nociceptive responses by acting on A1 receptors. By using selective adenosine agonist on the evoked potential record of the rat spinal cord, the adenosine A1 receptor agonist obviously inhibited the slow ventral root potential, which is the C-fiber-evoked excitatory response associated with nociceptive information[2]. In the present study, ICV administration of R-PIA produced a very short period (15 min) of antinocicep-tion. Systemically administered adenosine analogue R-PIA did not affect synaptic neurotransmission in the hippo-campus, which are enriched in A1-type adenosine receptors, because they failed to reach the appropriate receptors[11]. Thus, it is likely with the tail flick test that the site of action of R-PIA is probably at the spinal cord, because ICV injection of R-PIA only produced fewer responsesin the tail flick test, although it has previously been reported that activity in the hot-plate test was observed[12]. In the hot-plate test, the potential for an additional supraspinal action would need to be considered.
Adenosine modulation of anesthesia has been extensively studied in the past[13]. The standard for determining anesthetic requirements is the MAC of an inhaled agent that prevents gross purposeful movement in response to a supramaximal painful stimulus. Because of its simplicity and reproducibility, the MAC concept has remained an important tool for studying anesthetic action. Previous studies have shown that on a halothane anesthetized rat the effect of adenosine in decreasing the halothane MAC was probably related to an adenosine-induced decrease in CNS noradrenergic transmission because noradrenergic neurotransmission was decreased following R-PIA administration in all brain regions[13]. The present results provided evidence that into R-PIA injection significantly reduced MAC of halothane by 29%, and this reduction in MAC of halothane was inhibited with pretreatment of A1 antagonist DPCPX suggests that R-PIA increased the effects of anesthetic suppression was directly mediated A1 receptor. However, ICV injection of R-PIA also exhibited antinociceptive action, and the relationships between neurotransmitter and adenosine decrease MAC of halothane are not very clear. It is important that the behavioral responses consist of nociception, motor responses and central processing were measured with MAC. We are still unable to dissect which element(s) are affected by the central action of R-PIA.
Agonists of adenosine A1 receptors (including R-PIA) have activation action on K+ channels in neurons of the CNS. ATP-sensitive K+ channel blocker could inhibit the antinociception of adenosine in mice[14]. Other responses induced by adenosine A1 receptor agonist also appear to be linked to the opening of K+ channels. Adenosine A1 receptors in cardiac and vascular muscle cells are coupled to ATP-sensitive K+ channels[4]. In the present study, the effect of R-PIA on reducing the MAC of halothane and antinociception were antagonized by pretreatment with voltage-gated K+ channel blocker 4-AP. The apparent involvement of voltage-gated K+ channels in R-PIA-induced effect, as our results suggest, was not unexpected because 4-AP, as a potent voltage-gated K+ channel blocker, blocks outward conducting potassium channels, thereby lowering the threshold for initiation of action potentials as well as prolonging action potential duration in excitable membrane. In addition, the antinociceptive effect of GABAB receptor agonist baclofen and [D-ala2]-deltorphin II was also antagonized by 4-AP[15]. K+ channel opener could potentiate the analgesic effect of morphine[16], and the spinal antinociceptive action of morphine was caused by the release of adenosine and subsequent activation of adenosine receptors within the spinal cord[6]. Taken together, it is possible that adenosine affected the A1 receptor, resulted in increased K+ conductance preventing pain signal transmission to produce spinal antinociception and ICV methods reducing the requirement of halothane. Thus voltage-gated K+ channels play a key role in the R-PIA-induced effect both by IT and ICV administration.
In summary, the results provide implications for the use of adenosine analog in anesthetic settings and pain management. As evidenced from our data, the use of an adenosine analog during painful stimuli and halothane anesthesia profoundly decreased the response to nociception and increased the anesthetic depression through the A1 receptor subtype, and its mechanisms involves K+ channel activation. This observation pertains during other forms of volatile anesthesia and other types of pain treatment although definitive conclusions require further investigation.
Footnote
Project supported by Young Teacher Research Grants from Jilin University and by Sciences and Technology Commission of Changchun (N
References
- Zetterstrom T, Vernet L, Ungerstedt U. Purine levels in the intact rat brain: studies with an implanted prefused hollow fiber. Neurosci Lett 1982;29:111-5.
- Nakamura I, Ohta Y, Kemmotsu O. Characterization of adenosine receptors mediating spinal sensory transmission related to nociceptive information in the rat. Anesthesiology 1997;87:577-84.
- Sollevi A. Adenosine for pain control. Acta Anaesthesiol Scand 1997;59:135-6.
- Lerman BB, Belardinelli L. Antiadrenergic effects of adenosine on His-Purkinje automaticity: evidence for accentuated antagonism. Circulation 1991;83:1499-509.
- Huang QJ, Hao XL, Minor TR. Brain adenosine mediates interleukin-1β-induced behavioral depression in rats. Acta Pharm Sin 2002;34:421-5.
- Kekesi G, Dobos I, Benedek G. The antinociceptive potencies and interactions of endogenous ligands during continuous intrathecal administration: adenosine, agmatine, and endomorphin-1. Anesth Analg 2004;98:420-6.
- Ma HC, Dohi S, Wang YF. Antinociceptive and Sedative effects induced by carbachol and oxycodone into brainstem pontine reticular formation in rats. Anesth Analg 2001;92:1307-15.
- Zeng W, Dohi S, Shimonaka H. Spinal antinociceptive action of Na+-K+ pump inhibitor ouabain and its interaction with morphine and lidocaine in rats. Anesthesiology 1999;90:500-8.
- Ishizawa Y, Ma HC, Dohi S. Effects of cholinomimetic injection into the brain stem reticular formation on halothane anesthesia and antinociception in rats. J Pharmacol Exp Ther 2000;293:845-51.
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic Press; 1998. p 21–3.
- Bantel C, Tobin G, Li X. Intrathecal adenosine following spinal nerve ligation in rat: short residence time in cerebrospinal fluid and no change in A1 receptor binding. Anesthesiology 2002;96:103-8.
- Huang QJ, Jiang H, Hao XL, Minor TR. Brain IL-1β was involved in reserpine-induced behavioral depression in rats. Acta Pharmacol Sin 2004;25:293-6.
- Birch BD, Louie GL, Vickery RS. L-Phenylisopropyladeno-sine diminishes halothane requirements and decreases noradrenergic neurotransmission in rats. Life Sci 1988;42:1355-60.
- Maria O, Jose MB. Role of ATP-sensitive K+ channels in anti-nociception induced by R-PIA, an adenosine A1 receptor agonist. Nauyn-Schmiedeberg’s Arch Pharmacol 1994;350:57-62.
- Ong J, Kerr DI. Recent advances in GABAB receptors: from pharmacology to molecular biology. Acta Pharmacol Sin 2000;21:111-23.
- Asano T, Dohi S, Iida H. Antinociceptive action of epidural K+ (ATP) channel openers via interaction with morphine and α2-adrenergic agonist in rats. Anesth Analg 2000;90:1146-51.