
Estrogen stimulates release of secreted amyloid precursor protein from primary rat cortical neurons via protein kinase C pathway1
Introduction
Alzheimer’s disease (AD) is characterized by two major pathological lesions in the brain: intracellular neurofibrillary tangles and extracellular deposition of senile plaques composed mainly of amyloid-β protein (Aβ)[1]. Efforts have been made to explore the relative contribution of plaques and tangles to the pathogenesis of AD. In recent years, the amyloid hypothesis has been accepted by many researchers, who regard accumulation of Aβ in the brain as the primary factor driving AD pathogenesis[2,3]. Aβ is derived from a larger ubiquitous transmembrane protein, amyloid precursor protein (APP). APP is cleaved through at least two different pathways: α-secretase pathway and β-secretase pathway. Through the β-secretase pathway, which involves β-secretase and γ-secretase, APP is cleaved into two fragments: sAPPβ and Aβ. Alternatively, through the α-secretase pathway, which involves α-secretase and γ-secretase, APP is cleaved within the domain of amyloidogenic Aβ, thus precluding the generation of Aβ and producing non-amyloidogenic secreted APP (sAPPα) and p3 (Aβ 17-40/42). Several studies demonstrated that the increased activity of the α-secretase pathway led to a decrease in the activity of the β-secretase pathway[4].
Estrogen is considered as a neurotropic and neuro-protective agent[5]. Estrogen replacement therapy (ERT) in postmenopausal women is related with a reduced risk and delayed onset of AD[6]. It is therefore worthwhile investigating whether estrogen produces its neuroprotective effect through the regulation of APP processing[7].
There are several lines of evidence supporting this hypothesis. In cell culture, the addition of estrogen resulted in increase in the production of sAPPα and a decrease in Aβ[8,9]. In animal models, estrogen treatment prevented the accumulation of Aβ in the brain of guinea pigs and transgenic mutant APP/PS1-expressing mice[10,11]. It was suggested that the increased release of sAPPα by estrogen was mediated through the phosphorylation of extracellular-regulated kinase 1 and 2 (ERK1/2)[12]. However, the precise mechanism of the neuroprotective effects of estrogen remains to be further investigated. The purposes of the present study are to explore: (1) whether the effect of estrogen on sAPPα release is mediated through the membrane sites or the nuclear receptors; (2) whether the effect of estrogen depends on its molecular configuration specificity; (3) whether protein kinase C (PKC) is involved in the regulation of APP processing; and (4) whether classical estrogen receptor antagonist ICI 182 780 blocks the sAPPα secretion induced by estrogen.
Materials and methods
Cells and cell culture procedures Cortical neurons from 1-d postnatal Sprague-Dawley female or male rats were prepared as described previously[13]. Cells were plated in DMEM (Life Technologies, Gaithersburg, Germany) containing 10% fetal bovine serum (GIBCO) in poly-L-lysine-coated (0.1 g/L; Sigma, St Louis, USA) 100-mm dishes (approximately 1.8×107 cells per dish) under standard conditions (37 ºC, 5% CO2). On the following day, the medium was replaced with phenol red-free Neurobasal Medium (GIBCO), supplemented with B27 (GIBCO), which produced cultures with >99% neuronal composition[14]. The medium was replaced every 3 d. On the ninth day, the medium was replaced with phenol red-free Neurobasal Medium, supplemented with N2 (10% volume as described in guideline, GIBCO, Rockville, USA). Estrogen and/or other chemicals were added to the dishes. The chemicals used were 17β-estradiol (Sigma) dissolved in dimethyl sulfoxide (Me2SO), 17β-estradiol (Sigma) dissolved in Me2SO; β-estradiol 6-(O-carboxymethyl) oxime: BSA (Sigma) dissolved in PBS, β-estradiol 17-hemisuccinate: BSA (Sigma, St Louis, USA) dissolved in PBS, ICI 182, 780 (TOCRIS) dissolved in Me2SO, and PKC inhibitor calphostin C (Calbiochem, La Jolla, USA) dissolved in Me2SO. After a 12-h incubation, the medium was collected for measurement of sAPPα. The medium was then centrifuged at 4000×g for 10 min to remove the cellular debris. The cleared supernatant was concentrated with 30 kDa pore size Amicon Ultra (Millipore Co, Billerica,USA) for analysis. Cell monolayers were washed three times with ice-cold PBS and lysed in 1×SDS loading buffer with protease inhibitors for 10 min on ice. The cell lysates were boiled for 5 min and then centrifuged for 10 min at 14 000×g. The proteins were stored at -20 ºC.
Western blot analysis The protein concentrations were determined by BCA protein quantitative analysis kit (Shenergy Biocolor BioScience & Technology Company). An equal amount of protein from the medium was subjected to 7%–10% gradient SDS-poly-acrylamide gel electrophoresis (PAGE) (Amresco). After electrophoretic separation, proteins were transferred onto a PVDF membrane and the polyclonal antibody Rat A Beta (1:1000, Signet), which is specific for Ab 3-16, was used to detect sAPPα. The efficiency of transfer was confirmed by staining the membrane with Ponceau S. The Ponceau S stain was then removed from the membrane by washing in PBST. Cellular APP was detected using monoclonal antibody 22C11 (1:200, Roche), which is specific for the amino terminus of APP. The amount of β-actin on the same membrane, which was determined by polyclonal antibody β-actin (1:1000, Santa Cruz), was taken as control. Antibody binding was detected by counter-staining with horseradish peroxidase-conjugated antibodies (1:1000, Calbiochem) and visualized using an ECL-detection kit (Pierce). The relative intensity of immunoreactive bands on the exposed film was quantified by a computer-assisted densitometry program (Smart view, Life Science Research Products and System Engineering).
Statistics Data from sAPPα measurement were analyzed by one-way ANOVA followed by Tukey post hoc test (SPSS software). Student’s t-test was used to analyze the data of cellular APP content. P<0.05 was considered to be statistically significant. Each experiment was repeated three to four times to verify the reproducibility of the results.
Results
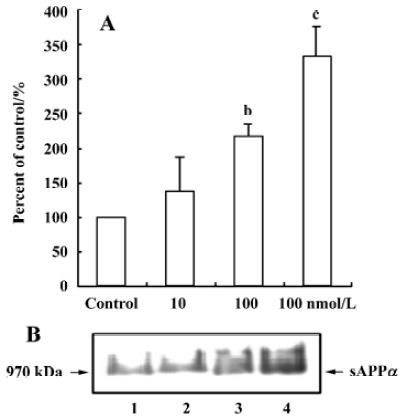
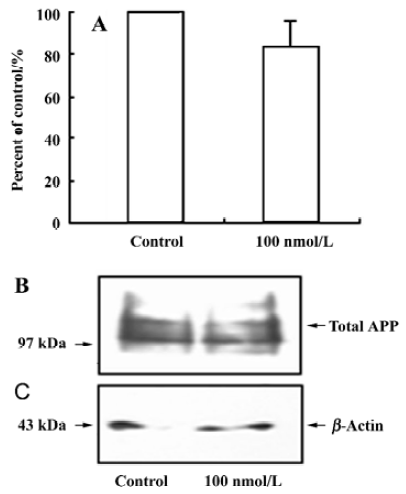
Effect of 17β-estradiol on sAPPα secretion and cellular APP content Primary cortical neurons were treated with 10, 100, and 1000 nmol/L 17β-estradiol. Figure 1 shows that a 12-h treatment with increasing concentrations of 17β-estradiol resulted in a significant dose-dependent increase in sAPPα release into the medium as compared with the sAPPα level in the control. 17β-Estradiol (100 nmol/L) did not alter the total cellular APP content in cultured cortical neurons (Figure 2).
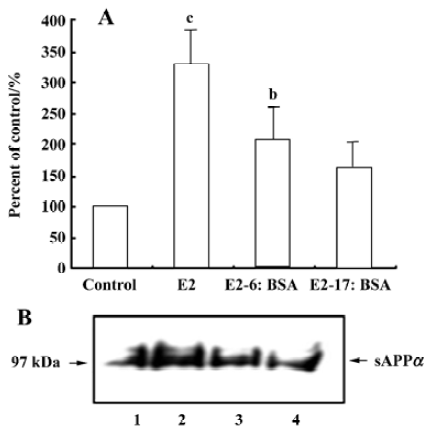
Effect of β-estradiol 6-(O-carboxymethyl) oxime: BSA on the release of sAPPα To investigate whether the membrane-impermeable estradiol-BSA conjugates, β-estradiol 6-(O-carboxymethyl) oxime: BSA and β-estradiol 17-hemi-succinate: BSA, exerted similar actions on sAPPα secretion as 17β-estradiol, neurons were incubated with these two agents. After a 12-h incubation, the supernatant was collected for measurement of sAPPα. β-Estradiol 6-(O-carboxymethyl) oxime:BSA (1000 nmol/L) increased sAPPα secretion, although the magnitude of the effect was smaller than that of 1000 nmol/L 17β-estradiol. Estradiol 17-hemisuccinate: BSA (1000 nmol/L) also stimulated sAPPα secretion, but the difference was not statistically significant (Figure 3).
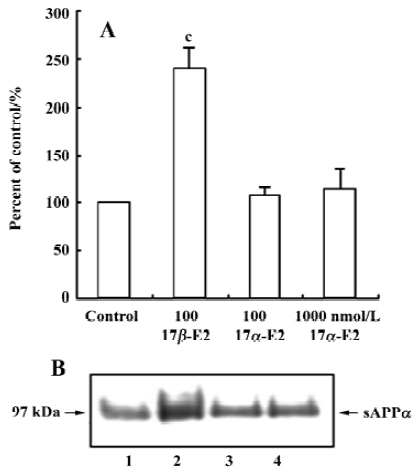
Effect of 17α-estradiol on the release of sAPPα In order to investigate whether estradiol stimulates sAPPα secretion depending on its configuration specificity, we treated the neurons with 100 nmol/L, 1000 nmol/L 17α-estradiol and 100 nmol/L 17β-estradiol. Twelve hours later, the supernatant was collected and concentrated for Western blot analysis. The results showed that, in contrast to 17β-estradiol, 17α-estradiol did not alter sAPPα secretion (Figure 4).
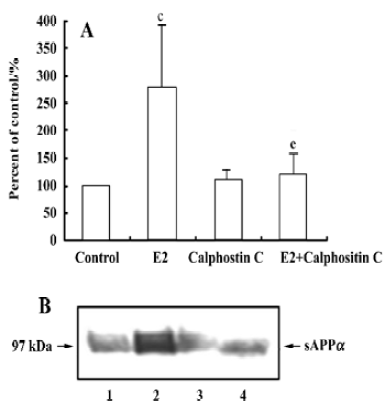
Effect of PKC inhibitor on 17β-estradiol-induced sAPPα secretion Calphostin C, a PKC inhibitor, was used to investigate the role of PKC on 17β-estradiol-induced secretion of sAPPα. Incubation with calphostin C (250 nmol/L) alone for 12 h did not change the sAPPα secretion. However, when the neurons were co-treated with calphostin C (250 nmol/L) and 17β-estradiol (100 nmol/L) for 12 h, the stimulatory effect of 17β-estradiol on sAPPα secretion disappeared (Figure 5).
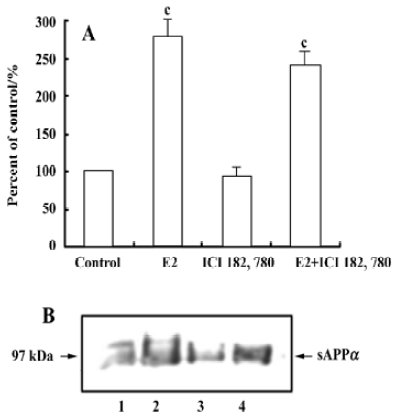
Effect of estrogen receptor antagonist on the 17β-estradiol-induced sAPPα secretion To explore whether activation of the classical estrogen receptors is involved in 17β-estradiol stimulating sAPPα secretion, rat cortical neurons were treated with 17β-estradiol (100 nmol/L), estrogen receptor antagonist ICI 182 780 (1 µmol/L) alone, and 17β-estradiol (100 nmol/L) together with ICI 182 780 (1 µmol/L). The results showed that ICI 182 780 alone produced no alteration in sAPPa secretion, while simultaneous administration of ICI 182 780 and 17β-estradiol resulted in a significant increase in sAPPα secretion, similar to the effect of 17β-estradiol (Figure 6).
Discussion
It appears that ERT prevents the onset of AD, but is not an effective treatment for AD[6]. However, recent clinical studies revealed that ERT brought beneficial as well as adverse effects[15]. Therefore, it is important to elucidate the mechanisms of the neuronal action of estrogen and to develop new drugs that retain the beneficial effects, but do not have the side effects of estrogen[16]. It is generally accepted that the effect of estrogen in preventing AD is mediated through regulating APP processing[2,7]. Our study provides evidence that estrogen increases sAPPα release without changing the total cellular APP content in cultured rat cortical neurons. Because the up-regulation of the α-secretase pathway mediating sAPPα production occurs within the same time course as down-regulation of the β-secretase pathway[4], it is reasonable to suggest that estrogen could reduce the generation of Aβ secondary to increases in sAPPα secretion and, in turn, exert its neuroprotective functions.
In addition to classical intracellular estrogen receptor, estrogen binding sites are also present on the cell membrane[5]. The actions of estrogen on its target tissues include a long-term “genomic” action mediated by intracellular estrogen receptors, and a rapid action through membrane binding sites that modulate a diversity of intracellular signal transduction pathways[17]. Therefore, it is interesting to know whether the estradiol-induced release of sAPPα is brought about through the mediation of nuclear or membrane binding sites. Our data clearly demonstrated that β-estradiol 6-(O-carboxy-methyl) oxime: BSA, a membrane-impermeable estradiol conjugate[18,19], produced a similar effect like estrogen to increase the release of sAPPα, suggesting the involvement of the membrane binding sites. Furthermore, the estrogen receptor antagonist ICI 182 780 did not affect the effect of estrogen, suggesting that the classical estrogen receptors ERα or ERβ were not involved in the above-mentioned estradiol effect. The role of classical estrogen receptors in the neuronal effect of estrogen is controversial. Some studies showed that the estrogen-induced rapid intracellular signal transduction cascades were unaffected by the classical estrogen receptor antagonists[18,20,21], such as tamoxifen and ICI 182 780, in cell lines that did or did not express estrogen receptors ERα and ERβ; while other studies showed that the rapid intracellular signal transduction was blocked by the classical estrogen receptor antagonists[13,22–24]. The reason for the difference in these studies is unknown, but it is probably a result of the different cells the researchers used and different parameters chosen for study, which appear through different membrane binding sites. The results of the present study suggest that the structure of the membrane binding sites responsible for the estradiol-induced sAPPα secretion is probably different from that of the classical estrogen receptors.
In present study, 17β-estradiol and its two BSA conjugates produced effects with different potencies. One of the possible explanations is that the combination of BSA alters the dimensional configuration of estradiol and thus changes its affinity for the membrane binding sites. In β-estradiol 6-(O-carboxymethyl) oxime: BSA, BSA attaches to estradiol at position 6, that is, on the “bottom” of the structure of estradiol, making the “top” (the C and D rings) of estradiol accessible. In contrast, in β-estradiol 17-hemisuccinate: BSA, BSA attaches to position 17 on the “top” of estradiol, making the “bottom” (the A and B rings) of the estradiol accessible. The difference in the configuration between the two conjugates can change the binding ability to the membrane sites and therefore change the effect in regulating sAPPα secretion. It is possible that a small amount of estradiol might dissociate from the conjugates, however, some studies reported that a very small amount of estradiol that dissociated from the conjugates did not exert effects on estrogen receptors[25].
Behl et al indicated that both 17β-estradiol and its isomer 17α-estradiol exhibited neuroprotective actions against oxidative stress[26]. 17α-estradiol does not bind to the nuclear ER, but 17α-estradiol was reported to stimulate phosphorylation of ERK1/2 in immature rat neurons[27]. Our present study showed that 17α-estradiol was unable to regulate the APP processing. Therefore, the mechanism for anti-oxidation effects of estrogens might be different from those of regulating APP metabolism.
Estrogen stimulates a rapid Ca2+ release from the intracellular calcium stores[28]. Yeon et al indicated that PKCε played an important role in modulating APP and that overexpression of the PKCεV1 region, which specifically binds to the receptor for activated C-kinase (RACK), blocked the phorbol ester-induced enhancement of sAPPα secretion[29]. Recently, Beyer et al reported that estrogen activated G-protein-coupled phospholipase C, leading to the release of Ca2+ from intracellular stores and the activation of PKC[30]. In the present study, we demonstrated that calphostin C prevented the increase of sAPPα secretion induced by 17β-estradiol, indicating the involvement of PKC.
In summary, our data indicate that, in cultured rat cortical neurons, estrogen stimulates sAPPα secretion through mechanisms involving membrane binding sites and the activation of PKC. The effects of estradiol are not mediated by classical ER. The details of the estrogen membrane binding sites and the related signaling pathways remain to be further studied.
Acknowledgement
We would like to express our sincere gratitude to Dr Xi-liang ZHA, Prof Yu HUANG, and Prof Li-min LU for their helpful suggestions and critical advices; and to Mr Zhen XU, Ms Bei FAN, and Yan-xia WANG for their assistance in Western blot performance.
Footnote
Project supported by the National Natural Science Foundation of China (N
References
- Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 1997;4:154-9.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002;297:353-6.
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992;256:184-5.
- Evin G, Weidemann A. Biogenesis and metabolism of Alzheimer’s disease Aβ amyloid peptides. Peptides 2002;23:1285-97.
- Garcia-Segura LM, Azcoitia I, DonCarlos LL. Neuroprotection by estradiol. Prog Neurobiol 2001;63:29-60.
- Owens CT. Estrogen replacement therapy for Alzheimer disease in postmenopausal women. Ann Pharmacother 2002;36:1273-6.
- Gandy S, Duff K. Post-menopausal estrogen deprivation and Alzheimer’s disease. Exp Gerontol 2000;35:503-11.
- Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE. Estrogen regulates metabolism of Alzheimer amyloid beta precursor protein. J Biol Chem 1994;269:13065-8.
- Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, et al. Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat Med 1998;4:447-51.
- Petanceska SS, Nagy V, Frail D, Gandy S. Ovariectomy and 17β- estradiol modulate the levels of Alzheimer’s amyloid β peptides in brain. Neurology 2000;54:2212-7.
- Zheng H, Xu H, Uljon SN, Gross R, Hardy K, Gaynor J, et al. Modulation of A (beta) peptides by estrogen in mouse models. J Neurochem 2002;80:191-6.
- Manthey D, Heck S, Engert S, Behl C. Estrogen induces a rapid secretion of amyloid precursor protein via the mitogen-activated protein kinase pathway. Eur J Biochem 2001;268:4285-91.
- Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci 1999;19:2455-63.
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosci Res 1993;35:567-76.
- Couzin J. Estrogen research. The great estrogen conundrum. Science 2003;302:1136-8.
- Wickelgren I. Estrogen research. Brain researchers try to salvage estrogen treatments. Science 2003;302:1138-9.
- Belcher SM, Zsarnovszky A. Estrogenic actions in the brain: estrogen, phytoestrogens, and rapid intracellular signaling mechanisms. J Pharmacol Exp Ther 2001;299:408-14.
- Beyer C, Karolczak M. Estrogenic stimulation of neurite growth in midbrain dopaminergic neurons depends on cAMP/protein kinase A signaling. J Neurosci Res 2000;59:107-16.
- Moats RK, Ramirez VD. Electron microscopic visualization of membrane-mediated uptake and translocation of estrogen-BSA: colloidal gold by hep G2 cells. J Endocrinol 2000;166:631-47.
- Morales A, Diaz M, Ropero AB, Nadal A, Alonso R. Estradiol modulates acetylcholine-induced Ca2+ signals in LHRH-releasing GT1-7 cells through a membrane binding site. Eur J Neurosci 2003;18:2505-14.
- Singh M, Setalo GJR, Guan X, Frail DE, Toran-Allerand CD. Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-alpha knock-out mice. J Neurosci 2000;20:1694-700.
- Mabuchi S, Ohmichi M, Kimura A, Nishio Y, Arimoto-Ishida E, Yada-Hashimoto N, et al. Estrogen inhibits paclitaxel-induced apoptosis via the phosphorylation of apoptosis signal-regulating kinase 1 in human ovarian cancer cell lines. Endocrinology 2004;145:49-58.
- Sato K, Matsuki N, Ohno Y, Nakazawa K. Estrogens inhibit l-glutamate uptake activity of astrocytes via membrane estrogen receptor alpha. J Neurochem 2003;86:1498-505.
- Guerra B, Diaz M, Alonso R, Marin R. Plasma membrane oestrogen receptor mediates neuroprotection against beta-amyloid toxicity through activation of Raf-1/MEK/ERK cascade in septal-derived cholinergic SN56 cells. J Neurochem 2004;91:99-109.
- Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology 1999;140:5455-8.
- Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, et al. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol 1997;51:535-41.
- Wong JK, Le HH, Zsarnovszky A, Belcher SM. Estrogens and ICI182, 780 (Faslodex) modulate mitosis and cell death in immature cerebellar neurons via rapid activation of p44/p42 mitogen-activated protein kinase. J Neurosci 2003;23:4984-95.
- Beyer C, Raab H. Nongenomic effects of oestrogen: embryonic mouse midbrain neurones respond with a rapid release of calcium from intracellular stores. Eur J Neurosci 1998;10:255-62.
- Yeon SW, Jung MW, Ha MJ, Kim SU, Huh K, Savage MJ, et al. Blockade of PKC epsilon activation attenuates phorbol ester-induced increase of alpha-secretase-derived secreted form of amyloid precursor protein. Biochem Biophys Res Commun 2001;280:782-7.
- Beyer C, Ivanova T, Karolczak M, Küppers E. Cell type-specificity of nonclassical estrogen signaling in the developing midbrain. J Steroid Biochem Mol Biol 2002;81:319-25.