
Caspase-1 inhibitor Ac-YVAD-CHO attenuates quinolinic acid-induced increases in p53 and apoptosis in rat striatum1
Introduction
Excitotoxin-induced degeneration of striatal neurons in amimals has been used as an animal model of Huntington’s disease (HD). Recent studies in transgenic animal models of HD demonstrated increased response of striatal neurons to excitotoxin, supporting a role of glutamate receptors in HD[1]. Excitotoxins acting on N-methyl-D-aspartate (NMDA) and kainic acid (KA) receptors will induce destruction of striatal GABAergic neurons by apoptotic mechanisms[2–4]. Caspases and tumor suppresser p53 play essential roles in apoptosis in a variety of cells including neurons[5–8]. Previous studies have reported that excitotoxin induces the activation of caspases and induces p53 expression[9–11]. Excitotoxin also reportedly activates nuclear factor-kappaB (NF-κB), but the mechanism by which this occurs remains unknown[12,13].
Recent studies have demonstrated that quinolinic acid (QA) induced significant increases in NF-κB binding activity in the nucleus[14]. NF-κB nuclear translocation mediates the upregulation of p53 and c-Myc in striatal neurons exposed to excitotoxic injury[15–17]. NMDA receptors activate NF-κB by selective degradation of an inhibitor protein, IκB-α. Excitotoxin-induced degradation of IκB-α involves a caspase-3-dependent mechanism in rat striatum and can be blunted by a caspase-3 inhibitor and a free radical scavenger[18,19]. In the present study we evaluated the role of caspase-1 inhibitor Ac-YVAD-CHO in QA-induced NF-κB activation, p53 induction, and apoptosis.
Materials and methods
Stereotaxic drug administration Sprague-Dawley rats (300–350 g) were obtained from the Experimental Animal Center of Soochow University (Certificate N
Isolation of genomic DNA and electrophoresis Striatal genomic DNA was prepared as described by Qin et al[3]. Briefly, striatal tissues were homogenized in a buffer containing NaCl 100 mmol/L, edetic acid 25 mmol/L, Tris-HCl 10 mmol/L (pH 8.0), 0.5% SDS, and RNase A 0.5 mg/L. Homogenates were incubated at 55 ºC for 2 h. Incubation was continued overnight after 0.6 mg protease K was added to the hoogenates. DNA was extracted with phenol:chlorform:isoamyl alcohol (24:25:1). DNA fragments were separated on 2% agarose gel and detected with a UV trasmilluminator after being stained with ethidium bromide.
Caspase-1 activity assay The caspase-1 activity assay was performed with an enzyme activity assay kit (Caspase-I/ICE Colorimetric Assay Kit, BioVision) according to the manufacturer’s instructions. Each striatal tissue was homogenized in 500 µL cell lysis buffer and centrifuged for 10 min at 10 000×g. The supernatant was transferred to a fresh tube and kept on ice. Protein concentration was determined using BCA kit (Pierce, Rockford, IL). For each 100 mg protein was diluted to 50 µL with cell lysis buffer and 50 µL 2×reaction buffer (containing DTT 10 mmol/L) was added, then 5 µL of the 4 µmol/L YVAD-pNA substrate (200 µmol/L) and the mixture was inculated at 37 ºC for 1 h. Read samples at 405 nm in a spectrophotometer (Bio-Rad Smart Spec 3000).
Western blot analysis Western blot analysis was performed as described previously[16]. Striatal tissues were homogenized in a buffer containing Tris-HCl 10 mmol/L (pH 7.4), NaCl 150 mmol/L, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, edetic acid 5 mmol/L, PMSF 1 mmol/L, aprotinin 0.28 kU/L, leupeptin 50 mg/L, benzamidine 1 mmol/L, pepstain A 7 mg/L. Protein concentration was determined using the BCA kit. Thirty micrograms of protein from each sample was subjected to electrophoresis on a 12% SDS-PAGE gel using a constant current. Proteins were transferred to nitrocellulose membranes and incubated with anti-p53 antibody (p53, p240, Santa Cruz, CA) in Tris buffered saline containing 0.2% Tween-20 (TBST) and 3% nonfat dry milk for 3 h. Membranes were washed and incubated with horseradish peroxidase-conjugated second antibody in TBST containing 3% nonfat dry milk for 1 h. Immunoreactivity was measured with enhanced chemoluminescent autoradiography (ECL kit, Amersham, Arlington Heights, IL) according to the manufacturer's instructions.
Electrophoresis mobility shift assay Striatal nuclear proteins were prepared as described by Qin et al[14]. Protein concentration was determined with the BCA kit (Pierce, Rockford, IL). Synthetic double-stranded NF-κB and AP-1 oligonucleotidyl probes were purchased from Promega (Madison, WI) and labeled with [32P]ATP using T4 polynucleotide kinase (Promega). Nuclear proteins (6–14 µg) were incubated with [32P]labeled probes (40 000 c/min) for 15 min at room temperature in the binding buffer (Promega). Reaction mixtures were electrophoresed on a 4.5% non-denaturing polyacrymide gel, then dried and exposed to x-ray film at –80 ºC with intensifying screens for 24 to 48 h. The results were quantitatively analyzed using an image analyzer (SigmaPlot Pro 4).
Results
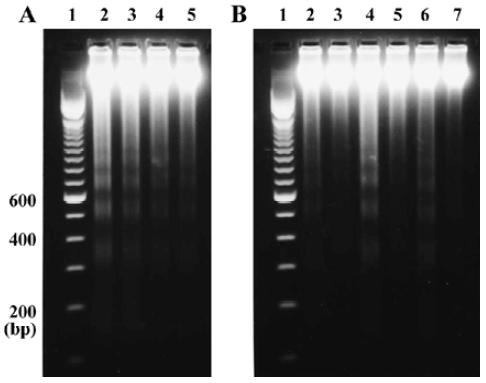
Effects of Ac-YVAD-CHO on QA-induced inter-nucleosomal DNA fragmentation To evaluate the consequences of caspase-1 inhibition on apoptosis, we studied the effect of Ac-YVAD-CHO on QA-induced internucleosomal DNA fragmentation. The results showed that QA (60 nmol) induced intense internucleo-somal DNA fragmentation 24 h after drug administration. The DNA fragments were generally multimers of 180–200 base pairs, indicating internucleo-somal DNA digestion by an endonuclease. QA-induced DNA fragmentation was strongly attenuated by Ac-YVAD-CHO in a dose-dependent manner (Figure 1A). The results also showed that Ac-YVAD-CHO (4 µg) inhibited QA-induced DNA fragmentation at all time points examined (12, 24, and 48 h after QA administration; Figure 1B).
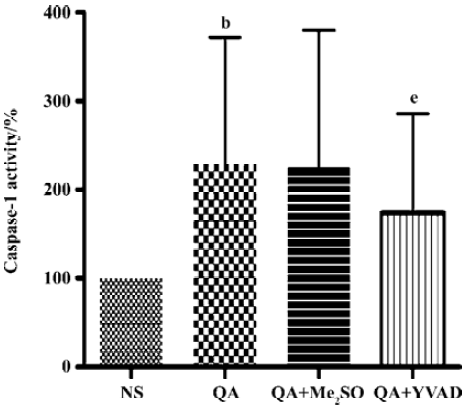
Effects of Ac-YVAD-CHO on a QA-induced increase in caspase-1 activity QA injection significantly activated caspase-1 activity in comparison with saline injected animals (P<0.05, n=6). The increase in caspase-1 activity induced by QA was significantly inhibited by Ac-YVAD-CHO (P<0.05, n=6); however, pre-treatment with Me2SO had no effect on the QA-induced activation of caspase-1 (Figure 2).
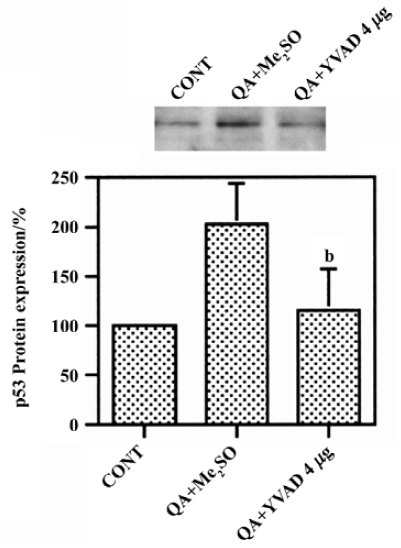
Effect of Ac-YVAD-CHO on QA-induced p53 induction QA induced significant increases in p53 protein levels by approximately 100%, which was significantly inhibited by Ac-YVAD-CHO (P<0.05, n=6, Figure 3).
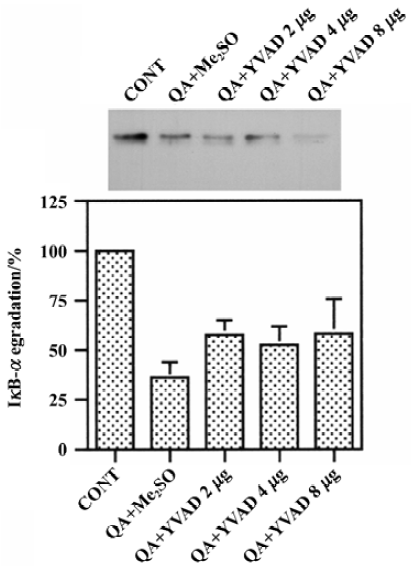
Effects of Ac-YVAD-CHO on QA-induced degradation of IκB-α QA treatment significantly reduced protein levels of IκB-α, indicating that IκB-α was degraded after QA treatment. Pre-treatment with various doses of Ac-YVAD-CHO (2–8 mg) had no significant effect on QA-induced degradation of IκB-α (Figure 4).
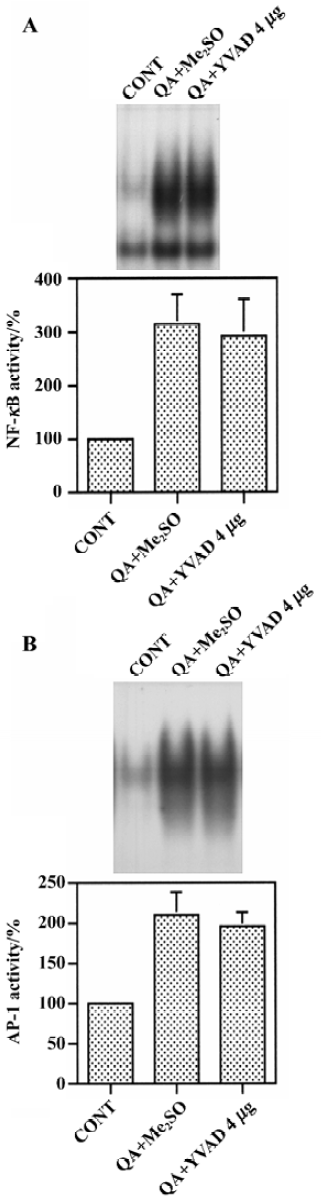
Effect of Ac-YVAD-CHO on the QA-induced activation of NF-κB and AP-1 Changes in NF-κB and AP-1 binding in nuclear extracts were measured by using an electrophoresis mobility shift assay. QA induced dramatic increases in NF-κB binding activity in nuclei. However, Ac-YVAD-CHO (4 mg) had no significant effect on QA-induced NF-κB activation (Figure 5A). Similarly, AP-1 binding activity was increased markedly after QA administration and Ac-YVAD-CHO failed to inhibit the QA-induced activation of AP-1 (Figure 5B).
Discussion
Apoptotic mechanisms are involved in the degeneration of strial neurons induced by the glutamate receptor agonist QA. In the present study, the role of caspase-1 in QA-induced p53 upregulation and NF-κB activation was investigated using the selective cell-permeable caspase-1 inhibitor Ac-YVAD-CHO. The results of the study showed that pre-treatment with Ac-YVAD-CHO dose-dependently inhibited QA-induced internucleosomal DNA fragmentation. Ac-YVAD-CHO had no significant effect on the QA-induced IκB-α degradation and NF-κB activation. However, Ac-YVAD-CHO partially inhibited QA-induced increase in p53 protein levels. These results suggest that caspase-1 plays a role in QA-induced p53 upregulation but not QA-induced IκB-α degradation.
It has been reported that glutamate receptor agonists activate NF-κB via the degradation of IκB-α[15,16]. Previous studies have further suggested that caspase-3, like protease, is involved in NMDA receptor-stimulated degradation of IκB-α[18,19]. Activation of NF-κB upregulates c-Myc and p53, indicating that NF-κB contributes to excitotoxin-induced apoptosis via the induction of cell cycle regulators[16,17,20]. p53 plays a critical role in cell death and survival. Its level is usually regulated by the rate of its degradation[21]. Now studies show that the levels of p53 can be regulated by NF-κB[16,17,22]. In the present study, caspase-1 inhibitor Ac-YVAD-CHO had no effect on either QA-induced IkB-α degradation or NF-κB activation. In contrast, Ac-YVAD-CHO significantly attenuated QA-induced increases in p53 protein levels and apoptosis. This study indicated that both NF-κB dependent and independent mechanisms are involved in the QA-induced upregulation of p53 and apoptosis.
In conclusion, we found that the caspase-1 inhibitor Ac-YVAD-CHO inhibited the QA-induced increase in p53 protein levels and internucleosomal DNA fragmentation, but had no effect on QA-induced IκB-α degradation and NF-κB activation. These results suggest that caspase-1 plays an important role in QA-induced p53 induction and apoptosis, but caspase-1 does not contribute to the QA-induced degradation of IκB-α or NF-κB activation.
Footnote
Project supported by the National Natural Science Foundation of China (N
References
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 2002;33:849-60.
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell culture. Proc Natl Acad Sci USA 1995;92:7162-6.
- Qin ZH, Wang Y, Chase TN. Stimulation of NMDA receptors induces apoptosis in rat brain. Brain Res 1996;725:166-76.
- Simonia NA, Getz RL, Leveque JC, Konradi C, Coyle JT. Kainate induces apoptosis in neurons. Neuroscience 1996;74:675-83.
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, et al. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol 1996;135:1085-96.
- Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, et al. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci 1997;17:553-62.
- Eldadah BA, Yakovlev A, Faden AI. The role of CED-3-related cysteine proteases in apoptosis of cerebellar granule cells. J Neurosci 1997;17:6105-13.
- Jordan J, Galino MF, Prehn JHM, Weichselbaum RR, Beckett M, Ghadge GD, et al. p53 expression induces apoptosis in hippocampal pyramidal neurons cultures. J Neurosci 1997;17:1397-405.
- Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA 1994;91:7525-9.
- Hughes PE, Alexi T, Yoshida T, Schreiber SS, Knusel B. Excitotoxic lesion of rat brain with quinolinic acid induces expression of p53 messenger RNA and protein and p53-inducible genes Bax and GADD-45 in brain areas showing DNA fragmenta-tion. Neuroscience 1997;74:1143-60.
- Du Y, Bales KR, Dodel RC, Hamilton-Byrd E, Horn JW, Czilli DL, et al. Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc Natl Acad Sci USA 1997;94:11657-62.
- Guerrini L, Blasi F, Denis-Donini S. Synaptic activation of NF-κB by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci USA 1995;92:9077-81.
- Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Stimulation of ionotropic glutamate receptors activate transcription factor NF-κB in primary neurons. Proc Natl Acad Sci USA 1995;92:9618-22.
- Qin ZH, Wang Y, Nakai M, Chase TN. Nuclear factor-κB contributes to excitotoxin-induced apoptosis. Mol Pharmacol 1998;53:33-42.
- Nakai M, Qin ZH, Chen JF, Wang Y, Chase TN. Kanic acid-induced apoptosis is associated with NF-κB activation. J Neuro-chem 1999;74:647-58.
- Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN. NF-κB nuclear translocation up-regulates c-Myc and p53 during N-methyl-D-aspartate receptor-mediated apoptosis. J Neurosci 1999;19:4023-33.
- Aleyasin H, Cregan SP, Iyirhiaro G, O’Hare MJ, Callaghan SM, Slack RS, et al. Nuclear factor-κB modulates the p53 response in neurons exposed to DNA damage. J Neurosci 2004;24:2963-73.
- Nakai M, Qin Z-H, Wang Y, Chase TN. NMDA and non-NMDA receptor-stimulated IκB-α degradation: differential effects of the caspase-3 inhibitor DEVD.CHO, free radical scanvenger OPC-14117 and ethanol. Brain Res 2000;859:207-16.
- Qin ZH, Wang Y, Chase TN. A caspase-3-like protease is involved in NF-κB activation induced by stimulation of N-methyl-D-aspartate receptors in rat striatum. Mol Brain Res 2000;80:111-22.
- Grilli M, Memo M. Possible role of NF-κB and p53 in the glutamate-induced pro-apoptotic neuronal pathway. Cell Death Differ 1999;6:22-7.
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997;88:323-31.
- Wu H, Lozano G. NF-κB activation of p53: a potential mechanism for supressing cell growth in response to stress. J Biol Chem 1994;269:20067-74.